La pseudopolyarthrite rhizomélique (PPR) se manifeste le plus souvent par une atteinte des ceintures scapulaire et pelvienne, associée à un syndrome inflam- matoire biologique. Elle peut être associée à une ACG ou être isolée. S’il existe un spectre de chevauchement entre ces 2 entités, les liens physiopathologiques n’ont pas encore été clairement élucidés.
Quelle fréquence ?
L’ACG survient également après 50 ans. C’est une pathologie plus rare ayant aussi une nette prédominance féminine. Elle est plus fréquente dans les pays nordiques. Le tabagisme est le principal facteur de risque environnemental. Bien que des études d’épidémiologie génétique aient établi un lien entre l’ACG et l’allèle HLA-DRB1*04, sa recherche n’a pas de place en pratique courante.
Corrélation entre les 2 pathologies : une PPR est retrouvée presque une fois sur deux dans l’artérite à cellules géantes. Quand la biopsie d’artère temporale est réalisée lors d’une PPR d’allure isolée, une ACG est découverte entre 41 % et 0 % des cas selon les séries.
Quand évoquer une PPR ?
Les douleurs touchent la ceinture scapulaire ± pelvienne : rachis cervical, épaules et racine des membres supérieurs ; rachis lombaire, région trochantérienne, hanches et racine des membres inférieurs. Les épaules sont presque constamment atteintes alors que le rachis cervical et la ceinture pelvienne ne le sont pas toujours (50-90 % des patients).
Elles sont volontiers bilatérales. L’horaire de survenue est typiquement inflammatoire : intensité maximale le matin au réveil, diminuant avec l’activité physique dans la journée ; les patients décrivent des douleurs les réveillant la nuit, vers 3-4 heures du matin. Un enraidissement est fréquent. La raideur matinale est parfois tellement invalidante que les malades ont besoin d’aide pour sortir de leur lit, alors qu’après un dérouillage souvent long (plus de 30 minutes), ils récupèrent une autonomie presque complète.
Les symptômes apparaissent généralement en quelques jours, mais peuvent aussi s’installer brutalement, du jour au lendemain. Les douleurs, parfois unilatérales, s’étendent rapidement aux 2 côtés. Une fébricule, voire une fièvre sont possibles et sont associées à une asthénie et une perte de poids chez presque la moitié des patients. Il s’agit parfois des premiers symptômes de la maladie. En revanche, une fièvre importante et prolongée doit faire rechercher une ACG et un diagnostic différentiel (infection, cancer).
L’interprétation de l’examen clinique doit tenir compte de l’heure à laquelle celui-ci est réalisé : la mobilisation active des membres peut être limitée dans la matinée mais normale plus tard dans la journée. La palpation des masses musculaires, du rachis cervical, lombaire bas et la mobilisation passive sont souvent douloureuses, sans signe inflammatoire.
Des arthrites périphériques peuvent être associées, touchant le plus souvent les poignets et les genoux. Elles doivent toujours faire évoquer des diag- nostics différentiels, en premier lieu la polyarthrite rhumatoïde (PR ;
Manifestations cliniques d’une ACG
Toutefois, chez certains patients, le seul point d’appel clinique est une altération de l’état général : fièvre traînante, claudication vasculaire ou complication ischémique de la maladie. Ces signes sont plus ou moins intenses, corrélés à la durée d’évolution de la maladie. Une toux d’apparition récente peut également être révélatrice. Un tableau clinique de PPR est associé dans environ 50 % des cas. Il n’a pas de spécificité par rapport à celui témoignant d’une PPR isolée.
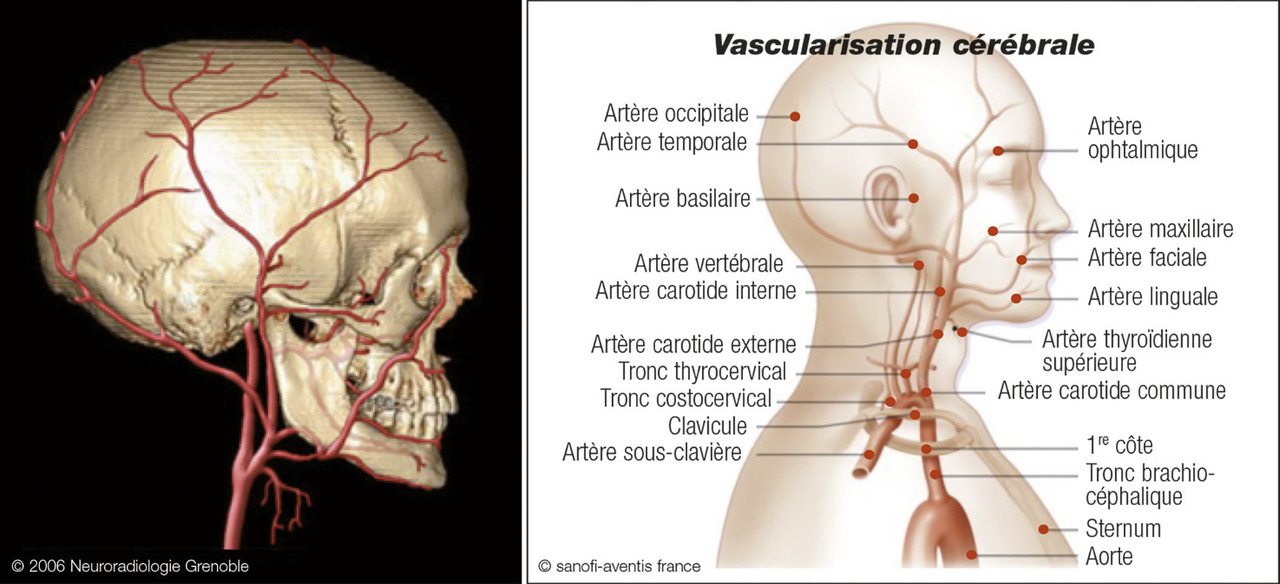
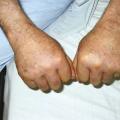
Les céphalées sont souvent temporales, unilatérales, à prédominance nocturne et matinale, mais elles peuvent prendre des formes différentes et sont parfois diffuses. Elles sont associées à une hyper- esthésie du cuir chevelu (classique mais inconstante) se traduisant par des douleurs déclenchées par le coiffage et le port des lunettes. Ces manifestations sont en lien avec l’atteinte de l’artère temporale (
L’atteinte de l’arc aortique peut entraîner un syndrome avec claudication des membres supérieurs, asymétrie tensionnelle et/ou de pouls, acrocyanose digitale. Celle des axes artériels des membres inférieurs peut être à l’origine d’une claudication mimant une artériopathie oblitérante banale. Des manifestations neuropsychiques, une paralysie oculomotrice, une sérite ou une neuropathie périphérique sont possibles mais rares (moins de 5 % des malades).
Les complications de l’ACG sont de nature essentiellement ischémique et largement dominées par les atteintes ophtalmologiques, qui concernent jusqu’à 20 % des patients. Il s’agit d’une cécité monoculaire brutale, voire bilatérale, parfois précédée de prodromes à type de flou visuel, scotome ou encore d’une diplopie (dans les cohortes historiques, une cécité définitive était observée chez 1 à 2 % des malades, monoculaire chez 2 à 5 % des patients). En cause : une neuropathie optique ischémique antérieure (NOIA ; dans la majorité des cas) ou postérieure, ou une occlusion de l’artère centrale de la rétine (OACR). Cause plus rare, un AVC dans le territoire vertébrobasilaire. Cette complication relativement fréquente (5 % des patients) survient surtout chez les sujets de sexe masculin ayant des facteurs de risque (majoritairement au niveau du territoire vertébrobasilaire).
Les complications d’une atteinte de l’aorte et/ou de ses branches, moins fréquentes, sont à redouter. Une aortite peut être à l’origine de la formation d’un anévrisme ou entraîner une dissection. Une atteinte valvulaire aortique est également possible, pouvant conduire à une insuffisance cardiaque justifiant dans certains cas une prise en charge chirurgicale lourde. Des infarctus du myocarde et mésentériques en lien avec l’atteinte artérielle inflammatoire sont également décrits.
Le diagnostic est aisé si la clinique est typique. Dans les formes avec signes généraux au premier plan, les diagnostics différentiels sont à évoquer au moment de planifier le bilan (pathologies systémiques, infectieuses et tumorales). Lorsque la maladie est révélée par une atteinte ischémique, un bilan cardiovasculaire exhaustif doit être conduit pour en éliminer les autres causes. Enfin, il n’est pas recommandé de réaliser une exploration systématique à la recherche d’une ACG devant une PPR isolée, sans atypie clinique et répondant à un traitement standard.
Faire le diagnostic
Dans l’immense majorité des cas d’ACG, le bilan biologique montre un syndrome inflammatoire important, avec une CRP ≥ 40 mg/L. Il peut s’accompagner d’une anémie, en général modérée, et d’une thrombocytose. Son absence, rare, doit systématiquement faire reconsidérer le diagnostic. Une cholestase anictérique est observée chez environ un tiers des patients, mais elle n’est ni sensible ni spécifique de la maladie. Aucun autre paramètre biologique n’est nécessaire. Le reste du bilan a donc pour but d’éliminer les diagnostics différentiels en fonction des symptômes, par exemple une vascularite systémique associée aux ANCA.
L’échographie doppler a une place de plus en plus importante dans le diagnostic de l’ACG, du fait de son accessibilité, de son faible coût, des données convaincantes et de sa bonne valeur prédictive (spécificité comprise entre 75 et 97 %). L’examen comporte une évaluation des artères temporales, de l’aorte et de ses branches. Un halo hypo-échogène circonférentiel de la paroi du vaisseau, dit « signe du halo », bilatéral, est très spécifique de la maladie (dans certains centres, on s’autorise à affirmer le diagnostic sans réaliser de biopsie d’artère temporale [BAT] lorsque la clinique est typique et que l’échographie doppler d’un opérateur entraîné montre cette atteinte bilatérale). L’examen peut également révéler des signes de sténose et/ou de thrombose artérielle. Des aspects similaires au signe du halo sont parfois retrouvés sur d’autres axes artériels, facilitant l’identification des formes moins typiques. Le TEP-scanner au 18-FDG met en évidence une hypercaptation intense du traceur au niveau de l’aorte et de ses branches chez environ 50 % des patients. Cet examen, aujourd’hui peu accessible, pourrait à l’avenir avoir une place dans le bilan lésionnel et le suivi de l’ACG, en particulier dans les formes extracrâniennes de la maladie. Parfois, une angio-TDM et/ou une angio-IRM sont utiles (par exemple, en cas de suspicion d’ACG compliquée d’un AVC dans le territoire vertébrobasilaire).
Une évaluation ophtalmologique est obligatoire chez tous les patients ayant eu des manifestations oculaires et souhaitable pour tous les malades atteints d’ACG. L’objectif est double : dépister des anomalies infracliniques et avoir une référence pour évaluer l’apparition de lésions ultérieures liées à une corticothérapie prolongée.
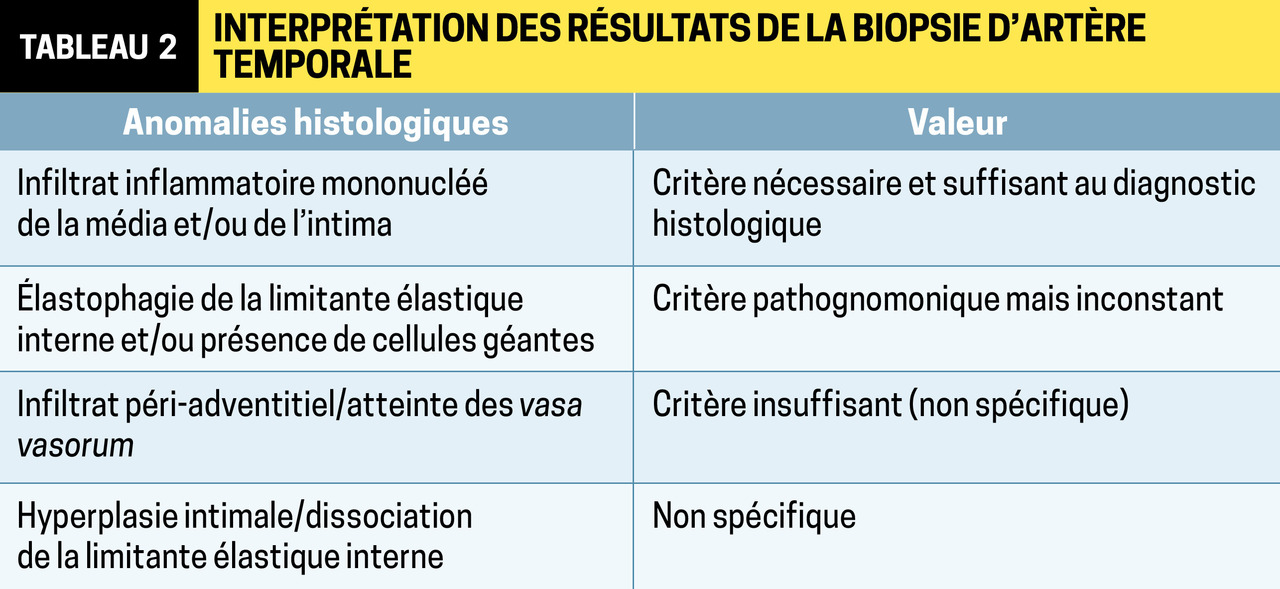
La BAT, examen clé du diagnostic, est un geste peu risqué, réalisé sous anesthésie locale. Le choix du côté à biopsier doit être guidé par les données cliniques ± échographiques. L’atteinte histologique classique est une panartérite segmentaire et focale (
PPR : d’abord soulager !
Les recommandations ACR/EULAR proposent de diminuer progressivement la corticothérapie après obtention d’une réponse clinicobiologique complète, afin d’arriver à une posologie de 10 mg par jour au plus tard 8 semaines après l’instauration du traitement, puis de la réduire de 1 mg tous les mois jusqu’à l’arrêt, en l’absence de rechute.
La décroissance des corticoïdes est parfois émaillée de rechutes de la maladie. L’augmentation au dernier palier efficace est alors recommandée. En cas de dépendance (sevrage impossible sans récidive) ou de résistance (persistance des symptômes malgré un corticoïde à dose habituellement efficace), une réévaluation par un spécialiste est nécessaire.
Il faut tout d’abord s’assurer qu’il s’agit bien d’une PPR. Si le diagnostic est confirmé, on peut envisager un traitement immunosuppresseur. Le plus utilisé est le méthotrexate, même si son efficacité est hétérogène. Le tocilizumab, anticorps monoclonal dirigé contre le récepteur de l’interleukine-6, est en cours d’étude dans les formes corticodépendantes (essai randomisé de phase III). Il a déjà montré une efficacité au début de la maladie pour diminuer la dose totale de corticoïdes reçus. La prescription de ces médicaments est réservée au spécialiste ; le tocilizumab ne peut être utilisé que dans le cadre d’essais cliniques.
ACG : prednisone avant tout
En dehors des situations d’urgence pouvant faire discuter le recours aux bolus intraveineux de méthyl- prednisolone et des formes particulières nécessitant une posologie initiale un peu plus élevée, le traitement d’attaque est la prednisone par voie orale, 0,7 mg/kg/j dans les ACG céphaliques non compliquées. Les formes résistantes sont exceptionnelles. Cette dose est en général maintenue pendant 2-4 semaines, dans tous les cas jusqu’à ce que les manifestations cliniques et les signes d’inflammation biologique soient amendés. Il n’y a pas de consensus concernant le protocole de décroissance mais il faut retenir les objectifs suivants (en l’absence de rechute) : 20 mg/j au 3e mois, 10 mg au 6e mois et 5 mg à 1 an du début du traitement. à partir de ce seuil de 5 mg débute la phase de sevrage – diminution très progressive, mensuelle, mg par mg – dont les objectifs sont d’accompagner le rétablissement de la sécrétion surrénalienne de cortisol endogène et éventuellement d’identifier la posologie minimale permettant d’obtenir une rémission prolongée de la maladie.
Les rechutes survenant lors de la décroissance et les récidives après arrêt du traitement sont fréquentes. Elles sont définies par la réapparition d’une symptomatologie clinique d’ACG, parfois différente du tableau initial, habituellement associée à un syndrome inflammatoire biologique. Leur contrôle requiert en général une posologie moindre de corticoïdes. Dans certains cas, le recours au palier précédent est suffisant, en particulier lorsqu’il s’agit d’une première rechute sans complication, survenant sous faible dose de prednisone.
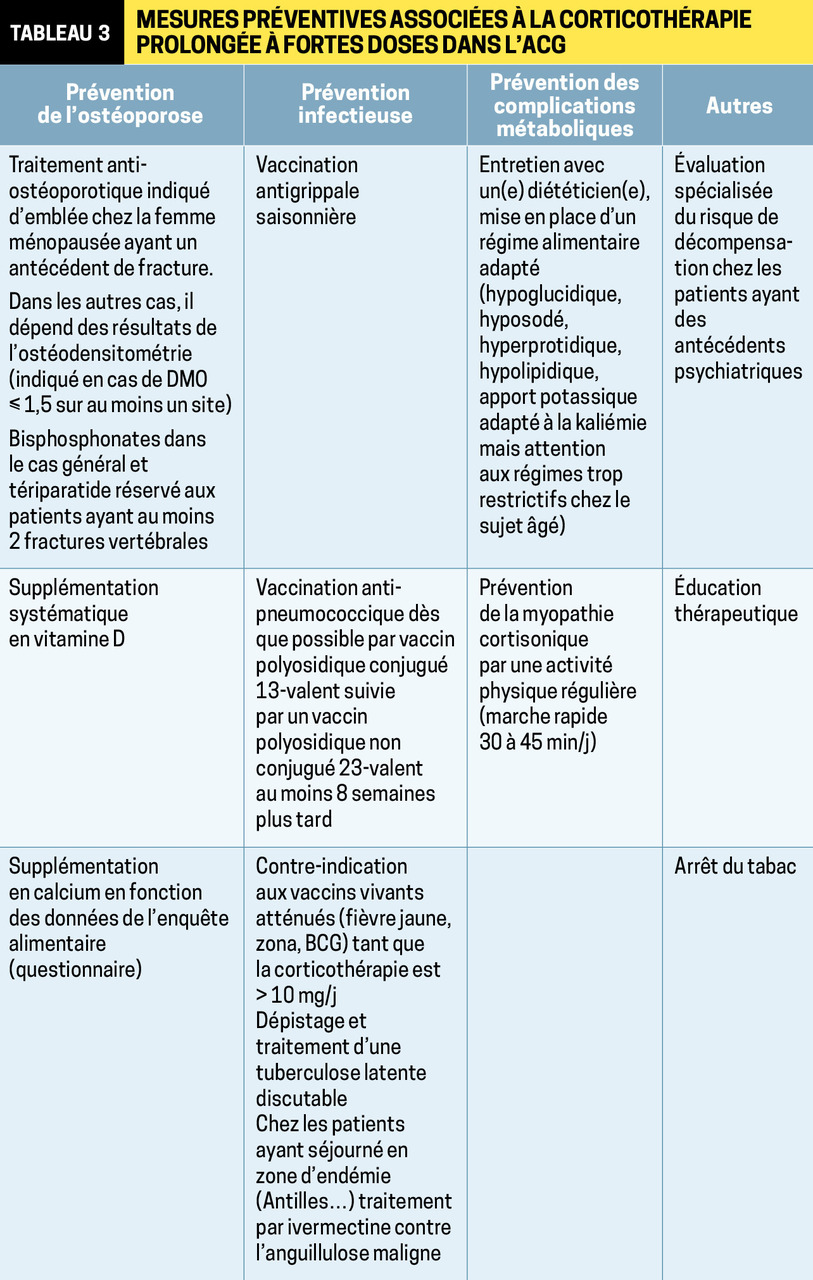
La corticothérapie est associée aux mesures préventives habituelles, qui doivent être strictes puisque la posologie cumulée reçue est en général très élevée : prévention de l’ostéoporose cortisonique, des infections, des effets indésirables métaboliques, évaluation psychiatrique chez les sujets à risque (
Un entretien avec un(e) diététicien(e) devrait être proposé à tous les patients. Une éducation thérapeutique est justifiée.
Concernant la prescription d’anticoagulants et antiagrégants plaquettaires, les publications sont discordantes. à ce jour, l’aspirine à dose antiagrégante doit être réservée aux patients ayant eu une complication ischémique, qu’elle soit oculaire ou non, afin de prévenir la récidive. Pour les sujets hospitalisés, une HBPM à dose préventive, bien qu’empirique, est la règle, d’autant plus s’il existe des signes généraux marqués. La prescription de statines doit se conformer aux recommandations habituelles (facteurs de risque cardiovasculaire classiques).
Les immunosuppresseurs/immunomodulateurs adjuvants sont pour le moment réservés aux rechutes multiples s’accompagnant d’une mauvaise tolérance de la corticothérapie et aux cas de corticodépendance prolongée à une dose supérieure à 7,5 mg/j. Ils ont pour objectif une épargne, voire l’interruption des corticoïdes. Deux molécules peuvent être envisagées : méthotrexate et tocilizumab. Ce dernier a récemment démontré une bonne efficacité (rémission clinique prolongée sans corticoïdes à 1 an). D’autres inhibiteurs de l’IL-6 sont en cours d’évaluation. La décision de débuter l’un de ces traitements est du ressort du spécialiste.
Surveillance et pronostic
La morbidité est importante, liée à la fois à l’artérite et à la corticothérapie prolongée. Les formes particulièrement graves et celles diagnostiquées trop tard peuvent avoir des conséquences lourdes (séquelles, décès). Cependant, il convient dans la très grande majorité des cas de rassurer les patients, car selon plusieurs études épidémiologiques la mortalité globale n’est que légèrement augmentée, voire non supérieure à celle de la population générale.
Pseudopolyarthrite rhizomélique : quel suivi ?
• Suivi de la douleur liée à la PPR : territoires douloureux, évaluation (EVA) par exemple sur les 7 derniers jours, réveils nocturnes liés à la douleur, durée du dérouillage matinal • Capacité d’élévation des membres supérieurs • Appréciation globale de la maladie par le patient à l’aide d’une EVA • Variation du poids • CRP et/ou VS • Arguments pour une éventuelle ACG • Arthrites périphériques, ténosynovites • Arguments pour un autre rhumatisme inflammatoire • Comorbidités interférant avec le traitement • Tolérance du traitement • Appréciation globale de la maladie par le médecin (EVA)
Buttgereit F, et al. Polymyalgia Rheumatica and Giant Cell Arteritis: A Systematic Review. JAMA 2016;315:2442-58.
HAS. Artérite à cellules géantes (Horton). Protocole national de diag- nostic et de soins. Août 2017.
Dans cet article
 Encadrés
Encadrés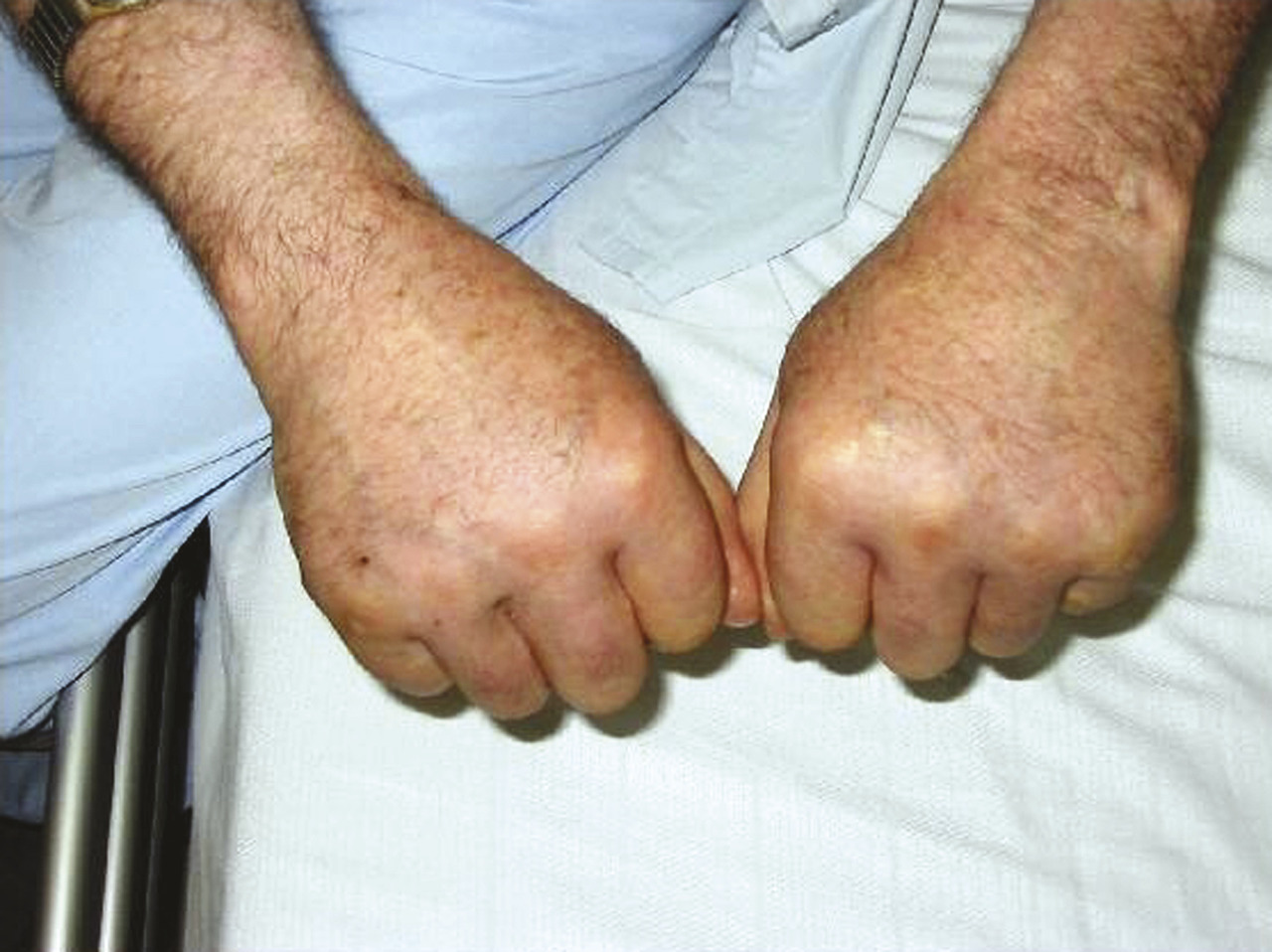





L’artérite à cellules géantes doit être évoquée devant des céphalées accompagnées d’un syndrome inflammatoire.
Une pseudopolyarthrite rhizomélique est associée dans 40 % des cas (mais peut être isolée).
La corticothérapie prolongée, pilier du traitement, est source de complications, à prévenir et dépister.