Les évoquer malgré une grande hétérogénéité clinique permet de prévenir certaines complications.
Il existe plus de 400 génodermatoses, maladies dermatologiques d’origine génétique, souvent orphelines. Traits communs : la rareté (prévalence < 2 000), une survenue souvent précoce (dès les premiers jours de vie ou l’enfance) et la chronicité.
Bulles et érosions post-bulleuses
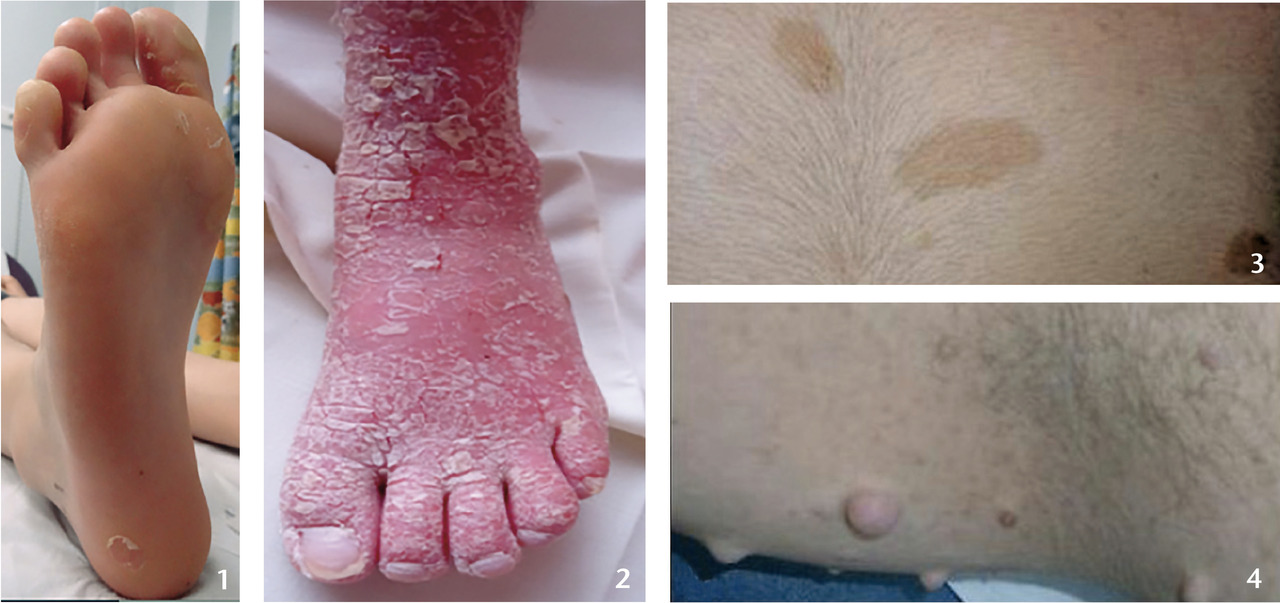
En période néonatale, de telles lésions incitent à la prudence. Elles peuvent révéler la fragilité cutanée d’une épidermolyse bulleuse. On évite l’application de tout adhésif risquant de majorer le décollement cutané. Les épidermolyses bulleuses héréditaires (EBH) résultent de mutations dans des gènes (19) codant des protéines de l’hémidesmosome, surtout. Cette structure assure la cohésion entre derme et épiderme. La fragilité cutanée est constatée régulièrement dès la naissance ; bulles et érosions post-bulleuses prédominent aux zones de frottement. Il y a souvent une atteinte muqueuse. Certains cas, moins sévères, se révèlent plus tardivement par des bulles des pieds et talons lors de la marche (fig. 1).1 La gravité est très variable, allant de formes localisées permettant une vie quasi normale à d’autres rapidement létales. Si la maladie est sévère, géné- ralisée, l’étendue des plaies, l’atteinte muqueuse, les cicatrisations à répétition sont sources de complications systémiques multiples : dénutrition, douleur, rétractions articulaires, syndrome inflammatoire chronique, amylose, carcinomes épidermoïdes cutanés.
Ichtyoses et troubles de la kératinisation
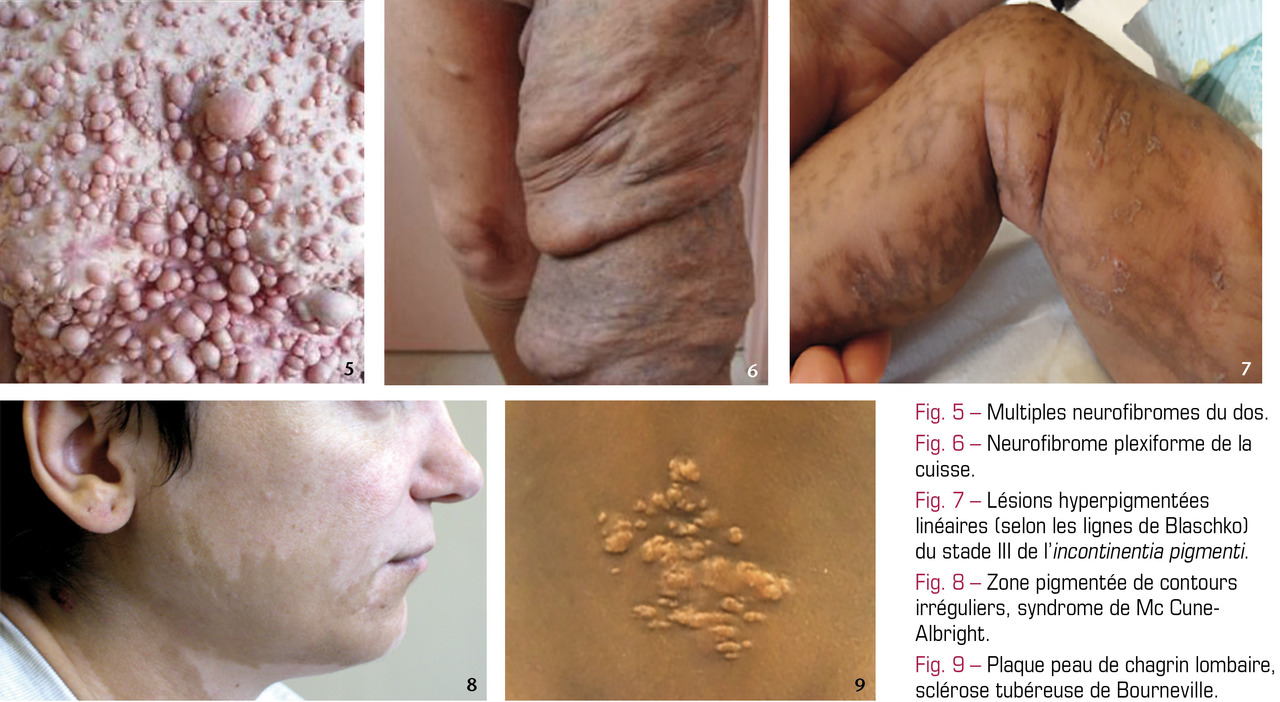
Ces maladies résultent d’une anomalie de la maturation épidermique (formation de la couche cornée). Cliniquement : squames épaisses (fig. 2), érythrodermie… Des manifestations extracutanées sont possibles, surtout neurologiques et ophtalmologiques. On est attentif au développement psychomoteur de l’enfant et au risque de carence en vitamine D.
L’ichtyose vulgaire (transmission autosomique dominante) apparaît rapidement et persiste toute la vie. Les squames sont fines, l’atteinte symétrique prédomine aux faces d’extension des membres et respecte en général les grands plis. Elle s’améliore spontanément l’été et s’aggrave pendant l’hiver. Le prurit est le plus souvent modéré.
L’ichtyose récessive liée à l’X se manifeste rapidement après la naissance. Les lésions concernent de manière symétrique les membres supérieurs et inférieurs et les faces latérales du visage et du tronc. En général, les grands plis sont moins respectés que dans l’ichtyose vulgaire, et les squames sont de plus grande taille. Les formes extensives donnent un aspect « sale » et inesthétique au tégument. Les femmes, souvent transmettrices, ont (pour seule lésion) une sécheresse cutanée de la face antérieure des jambes.
L’érythrodermie congénitale ichtyosiforme bulleuse est extrêmement rare. À la naissance, l’aspect « d’enfant ébouillanté » est rapidement remplacé par une érythrodermie couverte de larges décollements cutanés qui peuvent s’infecter. Plis et dos des mains et des pieds sont comme une « peau de serpent ». Lorsque l’enfant grandit, les décollements cutanés disparaissent pour laisser place à l’hyperkératose, souvent surinfectée et invalidante (odeur nauséabonde).
L’ichtyose vulgaire (transmission autosomique dominante) apparaît rapidement et persiste toute la vie. Les squames sont fines, l’atteinte symétrique prédomine aux faces d’extension des membres et respecte en général les grands plis. Elle s’améliore spontanément l’été et s’aggrave pendant l’hiver. Le prurit est le plus souvent modéré.
L’ichtyose récessive liée à l’X se manifeste rapidement après la naissance. Les lésions concernent de manière symétrique les membres supérieurs et inférieurs et les faces latérales du visage et du tronc. En général, les grands plis sont moins respectés que dans l’ichtyose vulgaire, et les squames sont de plus grande taille. Les formes extensives donnent un aspect « sale » et inesthétique au tégument. Les femmes, souvent transmettrices, ont (pour seule lésion) une sécheresse cutanée de la face antérieure des jambes.
L’érythrodermie congénitale ichtyosiforme bulleuse est extrêmement rare. À la naissance, l’aspect « d’enfant ébouillanté » est rapidement remplacé par une érythrodermie couverte de larges décollements cutanés qui peuvent s’infecter. Plis et dos des mains et des pieds sont comme une « peau de serpent ». Lorsque l’enfant grandit, les décollements cutanés disparaissent pour laisser place à l’hyperkératose, souvent surinfectée et invalidante (odeur nauséabonde).
Hyperpigmentations
Des taches café au lait (TCL) sont décrites dans diverses génodermatoses.2
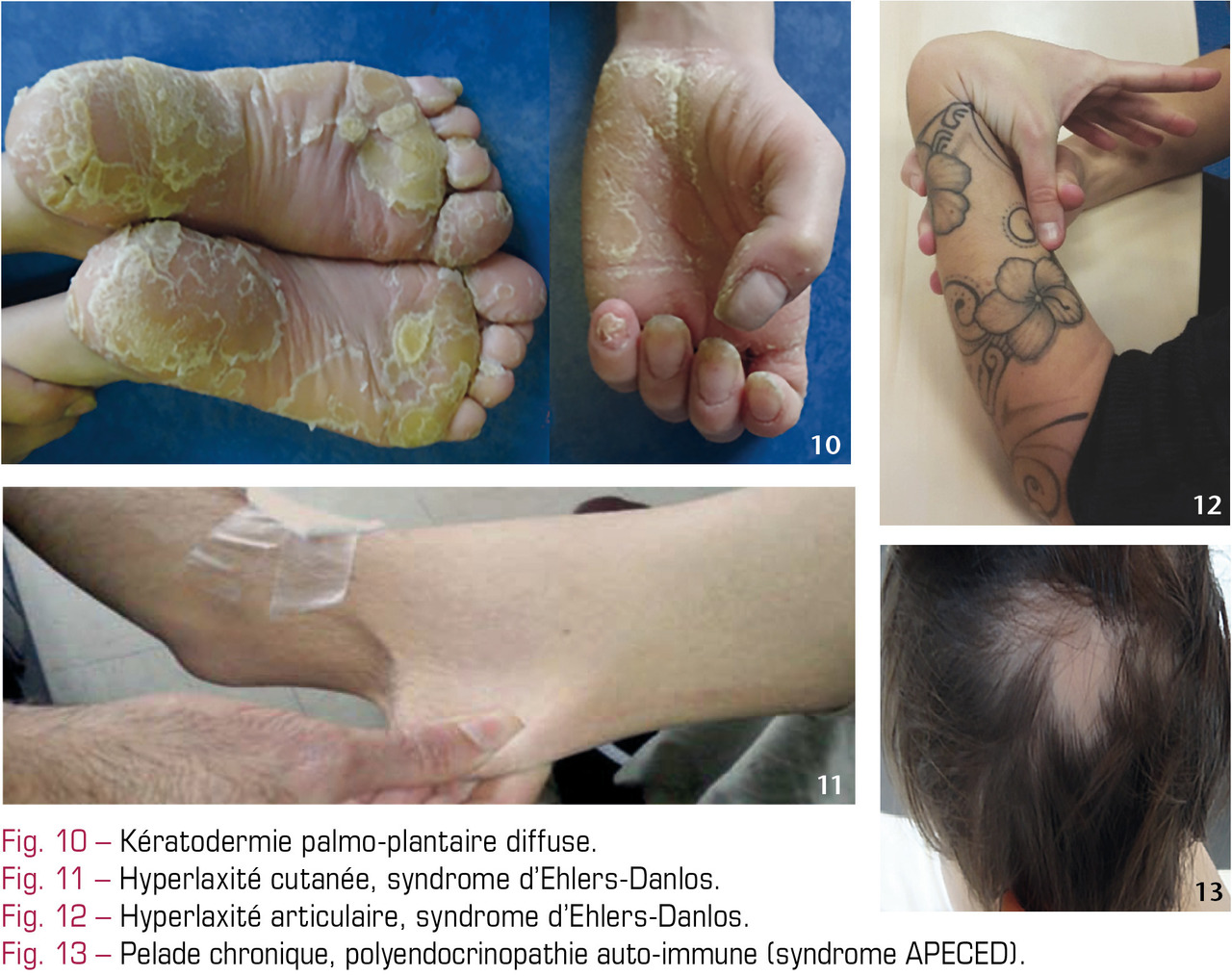
La plus fréquente, la neurofibromatose de type 1 (NF1) ou maladie de von Recklinghausen, autosomique dominante, a une incidence de 1/3 000 naissances dans le monde. Son expressivité inter- et intrafamiliale est variable. La neurofibromine, protéine codée par le gène NF1, régule la différenciation et la prolifération cellulaire. La mutation survient de novo dans 50 % des cas. Le diagnostic est clinique ; sa confirmation par analyse génétique n’est pas utile dans la plupart des cas.3 Critère le plus évident : plus de 6 taches café au lait (TCL) > 5 mm avant la puberté ou 1,5 cm après (fig. 3). Les TCL, en nombre croissant jusqu’à l’âge de 4 ans, sont des macules de couleur marron clair, arrondies/ovales, localisées surtout sur le tronc et les membres. Les lentigines (TCL millimétriques, 2e critère ; fig. 4), fréquentes, prédominent dans les régions axillaire et inguinale ; elles sont plus tardives. Les neurofibromes (NF), tumeurs cutanées papulo-nodulaires surviennent volontiers au moment de la puberté (fig. 5). Cas particuliers, le NF plexiforme (fig. 6), souvent unique, volumineux, de consistance molle, est congénital ou apparait précocement (première année de vie). La transformation en tumeur maligne des gaines nerveuses (TMGN) est possible. On surveille la croissance staturo-pondérale, la pression artérielle, les apprentissages, l’acuité et le champ visuel (gliome des voies optiques), la statique (scoliose par dysplasies vertébrales), l’audition et des signes orientant vers une puberté précoce.
Devant une hyperpigmentation linéaire et segmentaire (d’allure banale), il est essentiel de ne pas méconnaître les 2 affections suivantes dont les risques sont neurologiques, oculaires ou endocriniens.
Dans l’incontinentia pigmenti, atteignant surtout les nouveau-nés de sexe féminin (transmission autosomique dominante liée à l’X), les lésions sont linéaires (suivant les lignes de Blaschko) et évoluent souvent en 4 stades successifs parfois intriqués : vésiculo-bulleux, verruqueux hyperkératosique, hyperpigmenté (fig. 7) puis atrophique et hypopigmenté. En période néonatale, un avis dermatologique, neurologique et ophtalmologique urgent s’impose. Le risque de décollement de rétine et/ou de crise convulsive avoisine 20 %.
Le syndrome de McCune-Albright associe TCL, dysplasie fibreuse polyostosique (douleurs + hypertrophie volontiers asymétrique de la voûte crânienne et du massif facial) et manifestations endocriniennes secondaires à une hyperactivité en particulier gonadique (puberté précoce et aussi hyperthyroïdie, acromégalie, syndrome de Cushing, ostéomalacie hypophosphatémique). Les TCL, présentes dès l’enfance, sont de grande taille, à bords irréguliers (fig. 8), généralement limitées à un hémicorps. En cause : une mutation du gène GNAS responsable d’une activation de la voie AMPc associée (impliquée dans la signalisation de nombreuses hormones).
La plus fréquente, la neurofibromatose de type 1 (NF1) ou maladie de von Recklinghausen, autosomique dominante, a une incidence de 1/3 000 naissances dans le monde. Son expressivité inter- et intrafamiliale est variable. La neurofibromine, protéine codée par le gène NF1, régule la différenciation et la prolifération cellulaire. La mutation survient de novo dans 50 % des cas. Le diagnostic est clinique ; sa confirmation par analyse génétique n’est pas utile dans la plupart des cas.3 Critère le plus évident : plus de 6 taches café au lait (TCL) > 5 mm avant la puberté ou 1,5 cm après (fig. 3). Les TCL, en nombre croissant jusqu’à l’âge de 4 ans, sont des macules de couleur marron clair, arrondies/ovales, localisées surtout sur le tronc et les membres. Les lentigines (TCL millimétriques, 2e critère ; fig. 4), fréquentes, prédominent dans les régions axillaire et inguinale ; elles sont plus tardives. Les neurofibromes (NF), tumeurs cutanées papulo-nodulaires surviennent volontiers au moment de la puberté (fig. 5). Cas particuliers, le NF plexiforme (fig. 6), souvent unique, volumineux, de consistance molle, est congénital ou apparait précocement (première année de vie). La transformation en tumeur maligne des gaines nerveuses (TMGN) est possible. On surveille la croissance staturo-pondérale, la pression artérielle, les apprentissages, l’acuité et le champ visuel (gliome des voies optiques), la statique (scoliose par dysplasies vertébrales), l’audition et des signes orientant vers une puberté précoce.
Devant une hyperpigmentation linéaire et segmentaire (d’allure banale), il est essentiel de ne pas méconnaître les 2 affections suivantes dont les risques sont neurologiques, oculaires ou endocriniens.
Dans l’incontinentia pigmenti, atteignant surtout les nouveau-nés de sexe féminin (transmission autosomique dominante liée à l’X), les lésions sont linéaires (suivant les lignes de Blaschko) et évoluent souvent en 4 stades successifs parfois intriqués : vésiculo-bulleux, verruqueux hyperkératosique, hyperpigmenté (fig. 7) puis atrophique et hypopigmenté. En période néonatale, un avis dermatologique, neurologique et ophtalmologique urgent s’impose. Le risque de décollement de rétine et/ou de crise convulsive avoisine 20 %.
Le syndrome de McCune-Albright associe TCL, dysplasie fibreuse polyostosique (douleurs + hypertrophie volontiers asymétrique de la voûte crânienne et du massif facial) et manifestations endocriniennes secondaires à une hyperactivité en particulier gonadique (puberté précoce et aussi hyperthyroïdie, acromégalie, syndrome de Cushing, ostéomalacie hypophosphatémique). Les TCL, présentes dès l’enfance, sont de grande taille, à bords irréguliers (fig. 8), généralement limitées à un hémicorps. En cause : une mutation du gène GNAS responsable d’une activation de la voie AMPc associée (impliquée dans la signalisation de nombreuses hormones).
Hypopigmentations
es albinismes oculo-cutanés, le plus souvent de transmission autosomique récessive, sont caractérisés par une hypopigmentation ou absence de pigmentation de la peau, des phanères et des yeux. Ils résultent d’un déficit de production du pigment mélanique, par défaut biochimique de la mélanogenèse ou d’une anomalie du transport du pigment du mélanocyte vers le kératinocyte où il assure la protection du noyau. La prise en charge ophtalmologique est essentielle (photophobie, nystagmus). Des troubles de la coagulation (hématomes, épistaxis) et des infections à répétition doivent être recherchés pour éliminer des formes plus complexes. La photoprotection cutanée et oculaire est essentielle et s’accompagne d’une substitution en vitamine D à vie.
La sclérose tubéreuse de Bourneville (STB) se manifeste par la survenue de tumeurs bénignes (hamartomes) dans divers tissus. Elle résulte de mutations dans l’un des 2 gènes suppresseurs de tumeurs (TSC1 ou TSC2). La peau, le cerveau, les reins, les yeux et le cœur sont les organes à surveiller. Les macules hypopigmentées apparaissent dès les premiers mois de vie. Elles sont un critère diagnostique si leur nombre dépasse 3 et si la taille de chacune est supérieure à 0,5 cm. Leur aspect en « feuille de sorbier » est caractéristique, allongé, ovalaire, polygonal, asymétrique touchant avec prédilection le tronc et les fesses. L’examen cutané avec une lampe de Wood est utile. La plaque fibreuse du front et celle en peau de chagrin lombaire (fig. 9), caractéristiques, sont plus tardives. Les angiofibromes faciaux, très souvent confondus avec de l’acné juvénile, sont des papulo-nodules érythémateux télangiectasiques des régions périnasale et jugale. Leur précocité rectifie le diagnostic.4
L’hypopigmentation blashkolinéaire, généralement isolée, doit faire rechercher des anomalies extracutanées, surtout neurologiques (retard du développement, épilepsie).
La sclérose tubéreuse de Bourneville (STB) se manifeste par la survenue de tumeurs bénignes (hamartomes) dans divers tissus. Elle résulte de mutations dans l’un des 2 gènes suppresseurs de tumeurs (TSC1 ou TSC2). La peau, le cerveau, les reins, les yeux et le cœur sont les organes à surveiller. Les macules hypopigmentées apparaissent dès les premiers mois de vie. Elles sont un critère diagnostique si leur nombre dépasse 3 et si la taille de chacune est supérieure à 0,5 cm. Leur aspect en « feuille de sorbier » est caractéristique, allongé, ovalaire, polygonal, asymétrique touchant avec prédilection le tronc et les fesses. L’examen cutané avec une lampe de Wood est utile. La plaque fibreuse du front et celle en peau de chagrin lombaire (fig. 9), caractéristiques, sont plus tardives. Les angiofibromes faciaux, très souvent confondus avec de l’acné juvénile, sont des papulo-nodules érythémateux télangiectasiques des régions périnasale et jugale. Leur précocité rectifie le diagnostic.4
L’hypopigmentation blashkolinéaire, généralement isolée, doit faire rechercher des anomalies extracutanées, surtout neurologiques (retard du développement, épilepsie).
Kératodermies palmo-plantaires (KPP)
L’épaississement de la peau des paumes et des plantes (fig. 10) retentit sur la marche (souvent douloureuse) et la motricité fine (écriture). Dans certaines situations, elle peut révéler, associée à une alopécie, une cardiomyopathie sévère. Cette association est expliquée par la similitude des desmosomes assurant la cohésion entre kératinocytes et entre cardiomyocytes.5
Pathologies du tissu conjonctif
Le syndrome d’Ehlers-Danlos est l’expression d’une dysplasie des fibres collagènes. Dans la forme hypermobile (la plus fréquente), la peau est fine, hyperlaxe (fig. 11), avec des hématomes, des cicatrices atrophiques. S’y associe une hyperlaxité des grandes et petites articulations (fig. 12). Chez certains patients, l’absence de luxation articulaire et la discrétion de l’atteinte cutanée retardent le diagnostic. Signes évocateurs : fatigue, douleurs diffuses, fragilité cutanée et parfois troubles de la proprioception, de l’attention et de la mémoire. Contrairement à la forme vasculaire, il n’y a pas d’atteinte des gros vaisseaux pouvant mettre en jeu le pronostic vital. La transmission est surtout autosomique dominante. Le pronostic est essentiellement fonctionnel : le blocage des articulations retentit sur l’écriture et les gestes fins.
Dans la maladie de Marfan (dysplasie des fibres élastiques) l’atteinte est musculo-squelettique (grandes taille et envergure, déformation du thorax, hyperlaxité ligamentaire), oculaire (ectopie du cristallin) ou cardiovasculaire (dilatation aortique). L’examen dermatologique révèle des vergetures, des hernies (inguinales), une arachnodactylie (doigts longs et fins).
Dans la maladie de Marfan (dysplasie des fibres élastiques) l’atteinte est musculo-squelettique (grandes taille et envergure, déformation du thorax, hyperlaxité ligamentaire), oculaire (ectopie du cristallin) ou cardiovasculaire (dilatation aortique). L’examen dermatologique révèle des vergetures, des hernies (inguinales), une arachnodactylie (doigts longs et fins).
Pathologies dysimmunitaires
Une pelade (fig. 13) ou un vitiligo accompagnent parfois une polyendocrinopathie auto-immune. La répétition d’infections bactériennes ou mycosiques même banales conduit à éliminer un déficit immunitaire. Des lésions eczématiformes ou psoriasiformes atypiques sont souvent associées
références
1. Chiaverini C, Bourrat E, Mazereeuw-Hautier J, Hadj-Rabia S, Bodemer C, Lacour JP. Hereditary epidermolysis bullosa: French national guidelines (PNDS) for diagnosis and treatment. Ann Dermatol Venereol 2017;144:6-35.
2. Nunley KS, Gao F, Albers AC, Bayliss SJ, Gutmann DH. Predictive value of café au lait macules at initial consultation in the diagnosis of neuro-fibromatosis type 1. Arch Dermatol 2009;145: 883-7.
3. HAS, centre de référence labellisé neurofibromatoses. Protocole national de diagnostic et de soins (PNDS). Neurofibromatose 1. Décembre 2016.
4. Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:243-54.
5. Polivka L, Bodemer C, Hadj-Rabia S. Combination of palmoplantar keratoderma and hair shaft anomalies, the warning signal of severe arrhythmogenic cardiomyopathy: a systematic review on genetic desmosomal diseases. J Med Genet 2016; 53:289-95.
2. Nunley KS, Gao F, Albers AC, Bayliss SJ, Gutmann DH. Predictive value of café au lait macules at initial consultation in the diagnosis of neuro-fibromatosis type 1. Arch Dermatol 2009;145: 883-7.
3. HAS, centre de référence labellisé neurofibromatoses. Protocole national de diagnostic et de soins (PNDS). Neurofibromatose 1. Décembre 2016.
4. Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:243-54.
5. Polivka L, Bodemer C, Hadj-Rabia S. Combination of palmoplantar keratoderma and hair shaft anomalies, the warning signal of severe arrhythmogenic cardiomyopathy: a systematic review on genetic desmosomal diseases. J Med Genet 2016; 53:289-95.
Dans cet article


essentiel
➜ On examine le tégument et les muqueuses, mais aussi les annexes : cheveux, poils, glandes sudorales, dents.
➜ Le caractère affichant des lésions retentit sur les relations sociales, affectant la qualité de vie des patients et de leurs familles. Certaines peuvent mettre en jeu le pronostic vital.
➜ L’épiderme étant d’origine ectodermique, les manifestations extracutanées associées sont : neurologiques (macrocéphalie, épilepsie, trouble des apprentissages), HTA, ophtalmologiques, cardiaques et endocriniennes.
➜ Une à trois taches café au lait (TCL) isolées chez l’enfant d’âge scolaire ne sont pas pathologiques. En cas de TCL multiples, le risque de NF1 est estimé à 30 % et augmente à 75 % si le nombre est ≥ 6.