Il ne faut surtout pas manquer les formes entraînant la dysfonction d’un organe pouvant mettre en jeu le pronostic vital ou fonctionnel, et toujours évaluer et prendre en charge les signes d’inconfort persistants, qui peuvent retentir sévèrement sur la qualité de vie des patients.
La sarcoïdose est une maladie systémique, de cause inconnue, touchant presque constamment le poumon et le système lymphatique. Elle est caractérisée par la formation de granulomes tuberculoïdes dans les organes atteints.1-3 Le diagnostic et la prise en charge thérapeutique peuvent être difficiles en raison de la diversité des tableaux cliniques, du retentissement et de l’évolution de la maladie. Les objectifs du clinicien sont de ne pas manquer les formes avec dysfonction d’un organe pouvant mettre en jeu le pronostic vital ou fonctionnel, qualifiées de « dangereuses » mais aussi d’évaluer et de prendre en charge les signes d’inconfort persistants, qui peuvent retentir sévèrement sur la qualité de vie des patients. Le traitement de la sarcoïdose n’est pas systématique, mais si nécessaire, les corticoïdes, aux effets indésirables notables, sont à donner en première intention.
Épidémiologie
En France, la sarcoïdose est considérée comme une maladie rare, avec une prévalence estimée à près de 30/100 000 habitants et une incidence de 4,9/100 000 habitants/an dans une étude menée récemment en Seine-Saint-Denis.4 Les dernières études montrent que l’âge de déclaration de la maladie recule, apparaissant plus souvent après 45 ans chez les femmes et avant 45 ans chez les hommes.4-7 En revanche, la sarcoïdose est très rare avant l’âge de 15 ans et après 75 ans. Les femmes sont souvent un peu plus touchées, avec un pic d’incidence périménopausique. Le risque de développer une sarcoïdose est trois fois plus fréquent chez les patients d’origine africaine subsaharienne ou afro-américaine.4 L’obésité augmente le risque de sarcoïdose.8 La sarcoïdose est habituellement sporadique, mais des formes familiales se voient dans 4 à 8 % des cas. Enfin, le risque de survenue d’une sarcoïdose chez un parent du premier degré d’un patient atteint est multiplié par un facteur de 3,7.9 Le risque de transmission génétique a été calculé récemment à 39 %, les facteurs environnementaux apparaissant très importants.9 La sarcoïdose étant une maladie polygénique ne relève pas d’une consultation en conseil génétique.
Pathogénie
Le granulome sans nécrose caséeuse (contrairement à celui de la tuber culose) est la lésion histologique clef de la maladie sans pour autant en être spécifique. Le granulome est constitué d’un follicule central composé de macrophages, cellules épithélioïdes (macrophages activés produisant de nombreuses cytokines) et cellules géantes (fusion de cellules épithélioïdes) qui entrent en contact avec les lymphocytes T CD4+. Ce follicule est entouré d’une couronne lymphocytaire (T CD8+ et B). La cause de la maladie n’est pas connue, mais l’hypothèse principale est celle d’une réaction immunitaire exagérée (notamment de type Th1 avec la production de tumor necrosis factor alpha [TNFα], face à un antigène inconnu chez un patient génétiquement prédisposé, menant à la formation de granulomes.10 Un facteur environnemental est suggéré par la fréquence des localisations respiratoires, cutanées et ophtalmologiques, par la variabilité saisonnière de la maladie,11 par le pic d’incidence de cas de sarcoïdose multiviscérale noté chez les pompiers exposés aux nanoparticules après l’attaque du World Trade Center,12 par la présence de pathogènes (Propionibacterium acnes, peptides de mycobactéries) dans les tissus de certains patients,13 et par les variations importantes du nombre de cas dans certaines zones géographiques limitées.14 Les mécanismes expliquant la persistance du granulome et du développement de fibrose dans certains cas ne sont pas bien compris mais pourraient faire intervenir entre autres les lymphocytes T régulateurs, Th17.1, Th2 et les macrophages polarisés de type M2.10, 15
Circonstances de découverte
Délai diagnostique
Le diagnostic de sarcoïdose est parfois difficile à poser, avec un délai après les premières manifestations cliniques pouvant dépasser 6 mois dans 30 % des cas. Les délais sont augmentés en cas de symptômes respiratoires, qui sont peu spécifiques et orientent à tort vers d’autres maladies plus fréquentes, vers un asthme ou une bronchite chronique, ou en cas de manifestations rares. En revanche, une atteinte cutanée, plus suggestive, entraîne un diagnostic rapide. Dans la moitié des cas, les patients consultent plus de quatre médecins successifs avant que le diagnostic ne soit posé.16
Mode de révélation
Les principaux modes de révélation sont les symptômes respiratoires (toux sèche persistante, dyspnée plus rare, douleurs thoraciques liées à de volumineuses adénopathies intrathoraciques), le syndrome de Löfgren, les atteintes extrathoraciques fréquentes (cutanées, ganglionnaires périphériques, oculaires) et les signes généraux, notamment une fatigue souvent profonde.17, 18 Dans moins de 10 % des cas, la découverte est fortuite, sur une imagerie thoracique.17 Enfin, la maladie peut être révélée devant une hypercalcémie (surtout après exposition solaire estivale, prescription inappropriée de vitamine D ou en cas d’atteinte rénale).
Le syndrome de Löfgren est défini par l’association d’adénopathies intrathoraciques hilaires bilatérales avec un érythème noueux (surtout chez les femmes), ou une atteinte inflammatoire périarticulaire bilatérale isolée des chevilles (plus fréquente chez l’homme).19 Ce tableau a un bon pronostic dans 90 % des cas.
La fatigue est notée dans 70 % des cas de sarcoïdose (v. infra),20 parfois invalidante, avec nécessité d’arrêts de travail, indépendamment de la sévérité des atteintes viscérales de la maladie. Une fièvre est rarement présente, sauf en cas de syndrome de Löfgren, de syndrome de Heerfordt (parotidite bilatérale + uvéite + paralysie faciale périphérique), d’atteinte hépatique ou rénale, une fièvre devant toujours faire éliminer un diagnostic différentiel. De même, un amaigrissement significatif est rare, sauf en cas de syndrome de Löfgren ou d’atteinte multiviscérale du sujet âgé.
Le syndrome de Löfgren est défini par l’association d’adénopathies intrathoraciques hilaires bilatérales avec un érythème noueux (surtout chez les femmes), ou une atteinte inflammatoire périarticulaire bilatérale isolée des chevilles (plus fréquente chez l’homme).19 Ce tableau a un bon pronostic dans 90 % des cas.
La fatigue est notée dans 70 % des cas de sarcoïdose (v. infra),20 parfois invalidante, avec nécessité d’arrêts de travail, indépendamment de la sévérité des atteintes viscérales de la maladie. Une fièvre est rarement présente, sauf en cas de syndrome de Löfgren, de syndrome de Heerfordt (parotidite bilatérale + uvéite + paralysie faciale périphérique), d’atteinte hépatique ou rénale, une fièvre devant toujours faire éliminer un diagnostic différentiel. De même, un amaigrissement significatif est rare, sauf en cas de syndrome de Löfgren ou d’atteinte multiviscérale du sujet âgé.
Démarche diagnostique
Critères diagnostiques
La confirmation diagnostique repose surtout sur un tableau clinico-radiologique le plus souvent évocateur ou au moins compatible, la mise en évidence de granulomes typiques sans nécrose caséeuse, et sur l’exclusion des autres causes de granulomatose, en particulier la tuberculose.1 Il n’existe pas de marqueur spécifique de la maladie, mais un score diagnostique en cours d’étude répertoriant les différentes données cliniques, biologiques, radiologiques et histopathologiques trouvées au cours de la sarcoïdose, pourrait être un outil intéressant.21 Certaines situations ne nécessitent pas le recours à la biopsie, comme le syndrome de Löfgren, très spécifique de la sarcoïdose, ou la présence d’adénopathies médiastinohilaires bilatérales typiques asymptomatiques, mais sous réserve d’une surveillance bien programmée.
Stratégie de recherche des granulomes
La mise en évidence de granulomes typiques reste le plus souvent nécessaire, surtout si le tableau n’est pas habituel et/ou lorsqu’un traitement est envisagé.2, 3
Il faut privilégier, si la clinique le permet, les biopsies des lésions facilement accessibles, et qui sont « rentables » comme les lésions cutanées (excepté l’érythème noueux), les adénopathies périphériques (prélevées sous contrôle échographique pour les adénopathies superficielles),22 la biopsie des nodules conjonctivaux.
Compte tenu de la prévalence de l’atteinte pulmonaire, les prélèvements réalisés au cours d’une endoscopie bronchique ont un très bon rendement diagnostique et sont à envisager en l’absence de lésions superficielles. En outre, la fibroscopie bronchique permet la recherche de mycobactéries et renseigne sur l’existence ou non de lésions macroscopiques, en particulier de sténoses. Les biopsies bronchiques étagées (au moins 6) sont positives dans 30 à 100 % des cas selon l’absence ou la présence de lésions bronchiques macroscopiques.2 Le lavage broncho-alvéolaire montre une alvéolite lymphocytaire modérée (20-50 %) à lymphocytes T CD4+ dans 80 % des cas. Un rapport lymphocytes T CD4/CD8 supérieur à 3,5 est évocateur de sarcoïdose mais est insuffisant pour affirmer le diagnostic.3 Les biopsies pulmonaires transbronchiques ont une rentabilité diagnostique de 40 à 90 % mais exposent à un risque faible de pneumothorax ou d’hémoptysie. Si le patient a des adénopathies médiastinales et/ou hilaires, situation rencontrée dans 70 % des cas, la cytoponction transbronchique de ganglions intrathoraciques guidée par écho-endoscopie a une excellente rentabilité diagnostique et doit être privilégiée si les biopsies bronchiques sont négatives ; la présence du cytologiste sur place ne permet pas de diminuer la durée de l’examen mais de réduire le nombre de prélèvements.23 Cet examen a supplanté la médiastinoscopie.
En cas de négativité, on peut réaliser une biopsie des glandes salivaires accessoires (positive dans 40 % des cas dans les formes pauci-viscérales) particulièrement indiquée chez le sujet âgé ; une biopsie hépatique (sensibilité 70 % mais faible spécificité), réservée aux patients ayant une anomalie biologique hépatique ; ou un prélèvement orienté par la tomographie par émission de positons au 18F-fluorodésoxyglucose [18FFDG-TEP]). La biopsie transthoracique sous contrôle tomodensitométrique peut apporter le diagnostic en cas de nodule ou de condensation.
La biopsie pulmonaire chirurgicale par vidéochirurgie assistée est très exceptionnellement nécessaire et peut être discutée dans les formes atypiques : pneumopathie infiltrative diffuse sans adénopathie, ou de nodules pulmonaires. Elle tend à être supplantée par la réalisation de cryobiopsies pulmonaires transbronchiques sous anesthésie générale via une bronchoscopie rigide.24 Elle permet d’obtenir des biopsies de meilleure qualité (meilleure préservation tissulaire et de l’architecture) et de taille plus importante que les biopsies transbronchiques standard. Elle pourrait éviter le recours à la biopsie pulmonaire chirurgicale. Néanmoins, elle expose au risque d’hémoptysie et de pneumothorax.24
Il faut privilégier, si la clinique le permet, les biopsies des lésions facilement accessibles, et qui sont « rentables » comme les lésions cutanées (excepté l’érythème noueux), les adénopathies périphériques (prélevées sous contrôle échographique pour les adénopathies superficielles),22 la biopsie des nodules conjonctivaux.
Compte tenu de la prévalence de l’atteinte pulmonaire, les prélèvements réalisés au cours d’une endoscopie bronchique ont un très bon rendement diagnostique et sont à envisager en l’absence de lésions superficielles. En outre, la fibroscopie bronchique permet la recherche de mycobactéries et renseigne sur l’existence ou non de lésions macroscopiques, en particulier de sténoses. Les biopsies bronchiques étagées (au moins 6) sont positives dans 30 à 100 % des cas selon l’absence ou la présence de lésions bronchiques macroscopiques.2 Le lavage broncho-alvéolaire montre une alvéolite lymphocytaire modérée (20-50 %) à lymphocytes T CD4+ dans 80 % des cas. Un rapport lymphocytes T CD4/CD8 supérieur à 3,5 est évocateur de sarcoïdose mais est insuffisant pour affirmer le diagnostic.3 Les biopsies pulmonaires transbronchiques ont une rentabilité diagnostique de 40 à 90 % mais exposent à un risque faible de pneumothorax ou d’hémoptysie. Si le patient a des adénopathies médiastinales et/ou hilaires, situation rencontrée dans 70 % des cas, la cytoponction transbronchique de ganglions intrathoraciques guidée par écho-endoscopie a une excellente rentabilité diagnostique et doit être privilégiée si les biopsies bronchiques sont négatives ; la présence du cytologiste sur place ne permet pas de diminuer la durée de l’examen mais de réduire le nombre de prélèvements.23 Cet examen a supplanté la médiastinoscopie.
En cas de négativité, on peut réaliser une biopsie des glandes salivaires accessoires (positive dans 40 % des cas dans les formes pauci-viscérales) particulièrement indiquée chez le sujet âgé ; une biopsie hépatique (sensibilité 70 % mais faible spécificité), réservée aux patients ayant une anomalie biologique hépatique ; ou un prélèvement orienté par la tomographie par émission de positons au 18F-fluorodésoxyglucose [18FFDG-TEP]). La biopsie transthoracique sous contrôle tomodensitométrique peut apporter le diagnostic en cas de nodule ou de condensation.
La biopsie pulmonaire chirurgicale par vidéochirurgie assistée est très exceptionnellement nécessaire et peut être discutée dans les formes atypiques : pneumopathie infiltrative diffuse sans adénopathie, ou de nodules pulmonaires. Elle tend à être supplantée par la réalisation de cryobiopsies pulmonaires transbronchiques sous anesthésie générale via une bronchoscopie rigide.24 Elle permet d’obtenir des biopsies de meilleure qualité (meilleure préservation tissulaire et de l’architecture) et de taille plus importante que les biopsies transbronchiques standard. Elle pourrait éviter le recours à la biopsie pulmonaire chirurgicale. Néanmoins, elle expose au risque d’hémoptysie et de pneumothorax.24
Bilan initial
Le bilan à réaliser lors du diagnostic fait l’objet du tableau 1. Il s’attache à rassembler des arguments contre un diagnostic différentiel, à rechercher une localisation fréquente, potentiellement sévère et/ou asymptomatique et à confirmer une atteinte d’organe. Les principaux diagnostics différentiels sont la tuberculose ou les mycobactérioses non tuberculeuses, l’histoplasmose et la maladie de Whipple. Les pathologies environnementales et professionnelles sont aussi à exclure (bérylliose pulmonaire chronique) ainsi que les cancers s’accompagnant de granulomes (lymphomes, cancer pulmonaire). Enfin sont à éliminer les causes médicamenteuses ou une maladie dysimmunitaire : déficit immunitaire commun variable, reconstitution immunitaire chez les patients infectés par le virus de l’immunodéficience humaine (VIH), maladie de Crohn.
Bilan clinique
À l’interrogatoire, on recherche des éléments évocateurs de sarcoïdose (antécédent d’érythème noueux, antécédent familial de sarcoïdose), une exposition professionnelle (béryllium), ou environnementale à des aérocontaminants organiques, un contage tuberculeux ou un séjour en zone d’endémie, la notion d’infections à répétition (déficit immunitaire commun variable), la recherche d’une cause médicamenteuse de granulomatoses, en s’aidant du site www.pneumotox.org, les principaux médicaments à considérer étant les interférons α et γ, les anti-TNFα et les inhibiteurs du check-point immunitaire (PD-1, PD-L1). En cas de fatigue, le questionnaire Fatigue Assessment Scale (FAS) est un outil validé pour son évaluation et son suivi (tableau 2).25
L’examen clinique complet recherche des signes respiratoires, ophtalmologiques (baisse de l’acuité visuelle, œil rouge et douloureux), cardiaques (v. infra), neurologiques (v. infra), oto-rhino-laryngés (rhinite croûteuse persistante, épistaxis, obstruction nasale ou anosmie). L’atteinte laryngée qui peut être particulièrement sévère peut se traduire par une dysphonie, une dysphagie, une dyspnée inspiratoire et un stridor. La peau doit être examinée attentivement (v. infra). Toutes les aires ganglionnaires, dont les aires épitrochléennes, sont palpées. L’examen ophtalmologique spécialisé est systématique même en l’absence de symptôme.
Récemment, une large étude s’est intéressée au phénotype clinique de plus de 2 000 patients caucasiens dans 31 centres européens et a permis de différencier 5 groupes en fonction de l’atteinte prédominante et des associations d’organes : atteinte abdominale ; association d’atteintes oculaire, cardiaque, cutanée et du système nerveux central ; association d’atteintes cutanée et musculo-squelettique ; atteintes pulmonaire et ganglionnaire intrathoracique ; et atteinte extrapulmonaire.26
Il convient de rechercher des signes d’inconfort persistant (fatigue ; troubles des fonctions cognitives, notamment difficultés de concentration ; dépression ; atteinte des petites fibres nerveuses avec ou sans atteinte du système nerveux autonome ; douleurs diffuses) et d’évaluer le retentissement sur la qualité de vie.
L’examen clinique complet recherche des signes respiratoires, ophtalmologiques (baisse de l’acuité visuelle, œil rouge et douloureux), cardiaques (v. infra), neurologiques (v. infra), oto-rhino-laryngés (rhinite croûteuse persistante, épistaxis, obstruction nasale ou anosmie). L’atteinte laryngée qui peut être particulièrement sévère peut se traduire par une dysphonie, une dysphagie, une dyspnée inspiratoire et un stridor. La peau doit être examinée attentivement (v. infra). Toutes les aires ganglionnaires, dont les aires épitrochléennes, sont palpées. L’examen ophtalmologique spécialisé est systématique même en l’absence de symptôme.
Récemment, une large étude s’est intéressée au phénotype clinique de plus de 2 000 patients caucasiens dans 31 centres européens et a permis de différencier 5 groupes en fonction de l’atteinte prédominante et des associations d’organes : atteinte abdominale ; association d’atteintes oculaire, cardiaque, cutanée et du système nerveux central ; association d’atteintes cutanée et musculo-squelettique ; atteintes pulmonaire et ganglionnaire intrathoracique ; et atteinte extrapulmonaire.26
Il convient de rechercher des signes d’inconfort persistant (fatigue ; troubles des fonctions cognitives, notamment difficultés de concentration ; dépression ; atteinte des petites fibres nerveuses avec ou sans atteinte du système nerveux autonome ; douleurs diffuses) et d’évaluer le retentissement sur la qualité de vie.
Examens complémentaires
Un bilan biologique systématique comporte : hémogramme (une thrombopénie liée à la sarcoïdose peut résulter d’un hypersplénisme, d’une atteinte médullaire ou d’une auto- immunité),27 ionogramme sanguin, créatininémie et bandelette urinaire, calcémie, calciurie des 24 heures, électrophorèse des protéines plasmatiques (en cas d’hypogammaglobulinémie il faut éliminer un déficit immun commun variable), bilan hépatique (cholestase), sérologie pour le VIH. L’enzyme de conversion de l’angio- tensine n’est pas spécifique et peu sensible (élevée chez seulement 60 % des patients), mais une valeur supérieure à 2 fois la normale est très suggestive. Ses variations sont utiles pour suivre l’activité de la maladie, de même que la lymphopénie (prédominant sur les lymphocytes T CD4+), l’hypergammaglobulinémie (témoignant de l’activation des lymphocytes B) ou les anomalies du bilan calcique. L’hypercalcémie est la conséquence de la synthèse non freinable par les macrophages des granulomes, d’1-alpha-hydroxylase qui transforme la 25-hydroxyvitamine D3 en calcitriol. Dans 5 à 10 % des cas, il en résulte une hypercalcémie.
Les tests tuberculiniques, intradermoréaction ou interferon gamma release assays (IGRA), doivent être pratiqués. Ils montrent habituellement une anergie ou, rarement, peuvent être positifs, faisant discuter une tuberculose éventuellement associée, latente ou non. Enfin, une radiographie thoracique, des explorations fonctionnelles respiratoires (EFR) complètes avec spirométrie et mesure de la diffusion du monoxyde de carbone (DLCO), un électrocardiogramme (ECG) et une échocardiographie sont systématiquement réalisés (v. infra).
La tomodensitométrie thoracique n’est pas indispensable en cas de stade I typique sur la radiographie thoracique (adénopathies sans atteinte pulmonaire) mais s’avère généralement utile dans les autres cas, devenant indispensable en cas de tableau peu typique ou de suspicion d’une complication (v. infra). Le 18FFDG-TEP est rarement indiqué mais peut s’avérer très utile dans certains contextes très spécifiques3 car les cellules composant les granulomes ont un hypermétabolisme :28
– besoin d’identification d’un site occulte, possiblement biopsiable en l’absence de confirmation histologique aisée ;
– recherche d’une localisation cardiaque active lorsqu’une suspicion de localisation cardiaque est en suspens (examen réalisé dans un centre expert après un régime adapté, riche en lipides, pauvre en glucides pour supprimer la fixation métabolique myocardique du FDG) ;
– évaluation de l’activité associée à la présence d’une fibrose pulmonaire ;
– bilan d’une fatigue intense lors d’une maladie semblant non évolutive, avec des tests biologiques normaux pour préciser définitivement si une activité est encore présente, guidant l’initiation d’un traitement ;
– en cas d’hypertension pulmonaire, à la recherche d’un hypermétabolisme de ganglions médiastinaux devant une compression des vaisseaux pulmonaires.29
Les tests tuberculiniques, intradermoréaction ou interferon gamma release assays (IGRA), doivent être pratiqués. Ils montrent habituellement une anergie ou, rarement, peuvent être positifs, faisant discuter une tuberculose éventuellement associée, latente ou non. Enfin, une radiographie thoracique, des explorations fonctionnelles respiratoires (EFR) complètes avec spirométrie et mesure de la diffusion du monoxyde de carbone (DLCO), un électrocardiogramme (ECG) et une échocardiographie sont systématiquement réalisés (v. infra).
La tomodensitométrie thoracique n’est pas indispensable en cas de stade I typique sur la radiographie thoracique (adénopathies sans atteinte pulmonaire) mais s’avère généralement utile dans les autres cas, devenant indispensable en cas de tableau peu typique ou de suspicion d’une complication (v. infra). Le 18FFDG-TEP est rarement indiqué mais peut s’avérer très utile dans certains contextes très spécifiques3 car les cellules composant les granulomes ont un hypermétabolisme :28
– besoin d’identification d’un site occulte, possiblement biopsiable en l’absence de confirmation histologique aisée ;
– recherche d’une localisation cardiaque active lorsqu’une suspicion de localisation cardiaque est en suspens (examen réalisé dans un centre expert après un régime adapté, riche en lipides, pauvre en glucides pour supprimer la fixation métabolique myocardique du FDG) ;
– évaluation de l’activité associée à la présence d’une fibrose pulmonaire ;
– bilan d’une fatigue intense lors d’une maladie semblant non évolutive, avec des tests biologiques normaux pour préciser définitivement si une activité est encore présente, guidant l’initiation d’un traitement ;
– en cas d’hypertension pulmonaire, à la recherche d’un hypermétabolisme de ganglions médiastinaux devant une compression des vaisseaux pulmonaires.29
Sarcoïdose pulmonaire
La sarcoïdose peut atteindre tous les organes, néanmoins l’atteinte pulmonaire est la plus fréquente et présente dans plus 90 % des cas. Elle est, par ailleurs, la première cause de mortalité, principalement en cas de fibrose pulmonaire et/ou d’hypertension pulmonaire.30
L’auscultation pulmonaire est le plus souvent normale, contrastant avec les images radiologiques. Les râles crépitants sont notés dans 4 % des cas, des sibilants peuvent être audibles en cas d’atteinte bronchique. Une évolution anormalement aiguë de l’atteinte pulmonaire, une hémoptysie ou un hippocratisme digital doivent faire considérer un autre diagnostic.31
La radiographie thoracique est essentielle au diagnostic et pour le suivi de la maladie, elle est anormale dans 90 à 95 % des cas.32 Certains signes sont typiques et présents dans 50 à 80 % des cas : les adénopathies hilaires bilatérales symétriques et non compressives (fig. 1). La présence d’adénopathies médiastinales, sans atteinte hilaire ou unilatérale, est très atypique et doit faire rechercher un diagnostic différentiel (cancer, hémopathie, tuberculose). L’infiltration parenchymateuse, si elle est présente, prédomine dans les territoires supérieurs et moyens. La classification de Scadding distingue 5 stades corrélés avec la probabilité de guérison spontanée de la sarcoïdose :
– stade 0 : radiographie normale au cours d’une sarcoïdose extrathoracique ;
– stade I : stade le plus fréquent, caractérisé par des adénopathies sans atteinte parenchymateuse ;
– stade II : adénopathies associées à une atteinte parenchymateuse ;
– stade III : atteinte parenchymateuse isolée sans fibrose ;
– stade IV : fibrose pulmonaire.
La probabilité de guérison est respectivement de 60-90 %, 40-70 %, 10-20 % et 0 % pour les stades I, II, III et IV.
La tomodensitométrie thoracique a un grand intérêt diagnostique surtout en cas de présentation peu typique. Le tableau typique32 (fig. 2) est celui de l’association d’adénopathies médiastino-hilaires bilatérales et symétriques associées à des micronodules de distribution périlymphatique (les micronodules suivent les trajets lymphatiques et se retrouvent le long des axes péribronchovasculaires, à l’échelle du lobule secondaire dans les septums interlobulaires, en sous-pleural et notamment le long des scissures) prédominant dans les territoires supérieurs et moyens (fig. 3). Ce tableau permet de prédire avec plus de 95 % de sécurité un diagnostic de sarcoïdose chez 80 % des patients.2, 33, 34 Dans les rares cas où des lésions non habituelles prédominent, comme le verre dépoli ou les condensations, la présence d’adénopathies typiques ou de micronodules périlymphatiques même au second plan permet dans la plupart des cas de retenir le diagnostic de sarcoïdose.35, 36 La tomodensitométrie permet, en outre, de renseigner avec plus de précision sur le caractère réversible inflammatoire (micronodules, nodules) ou irréversible des lésions (fibrose). Les lésions de fibrose pulmonaire prédominent dans les territoires supérieurs moyens et périhilaires. Le tableau le plus typique est celui de réticulations avec distorsion péribronchovasculaire prédominant dans les lobes supérieurs dont le volume est réduit. Des adéno- athies calcifiées bilatérales peuvent témoigner de la déclaration ancienne de la maladie.37
Les EFR sont souvent normales ou discrètement anormales lors du diagnostic.18 Le retentissement fonctionnel consiste typiquement en un syndrome restrictif et un trouble de diffusion.2, 18 Le suivi de la capacité vitale forcée (CVF) est l’élément le plus fiable indispensable pour le suivi pulmonaire, avec la radiographie simple du thorax.32 Un trouble ventilatoire obstructif est présent dans 14 à 50 % des cas, et plusieurs mécanismes peuvent en être responsables.38 La DLCO est le plus souvent diminuée en raison d’une atteinte de la membrane alvéolocapillaire (par exemple en cas de fibrose) ou être le reflet d’une cause vasculaire (hypertension pulmonaire). Une mesure des débits inspiratoires est recommandée en cas de suspicion d’atteinte laryngée.
Le test de marche des 6 minutes peut mettre en évidence une réduction du parcours et une désaturation en oxygène et, chez les patients avec une dyspnée persistante sous traitement, ces anomalies conduisent à rechercher une hypertension pulmonaire. Une épreuve d’effort sur tapis ou ergocycle s’avère utile lorsque le mécanisme de la dyspnée n’est pas compris en orientant vers une cause pulmonaire, cardiovasculaire, musculaire spécifique et/ou un déconditionnement.
Certaines formes de sarcoïdose sont qualifiées de « dangereuses » car associées à un risque de mortalité, de morbidité significative, ou la nécessité d’un traitement lourd ou exceptionnel (biomédicament anti-TNFα, transplantation) : c’est le cas notamment de certaines atteintes pulmonaires, cardiaques et neurologiques.
L’auscultation pulmonaire est le plus souvent normale, contrastant avec les images radiologiques. Les râles crépitants sont notés dans 4 % des cas, des sibilants peuvent être audibles en cas d’atteinte bronchique. Une évolution anormalement aiguë de l’atteinte pulmonaire, une hémoptysie ou un hippocratisme digital doivent faire considérer un autre diagnostic.31
La radiographie thoracique est essentielle au diagnostic et pour le suivi de la maladie, elle est anormale dans 90 à 95 % des cas.32 Certains signes sont typiques et présents dans 50 à 80 % des cas : les adénopathies hilaires bilatérales symétriques et non compressives (fig. 1). La présence d’adénopathies médiastinales, sans atteinte hilaire ou unilatérale, est très atypique et doit faire rechercher un diagnostic différentiel (cancer, hémopathie, tuberculose). L’infiltration parenchymateuse, si elle est présente, prédomine dans les territoires supérieurs et moyens. La classification de Scadding distingue 5 stades corrélés avec la probabilité de guérison spontanée de la sarcoïdose :
– stade 0 : radiographie normale au cours d’une sarcoïdose extrathoracique ;
– stade I : stade le plus fréquent, caractérisé par des adénopathies sans atteinte parenchymateuse ;
– stade II : adénopathies associées à une atteinte parenchymateuse ;
– stade III : atteinte parenchymateuse isolée sans fibrose ;
– stade IV : fibrose pulmonaire.
La probabilité de guérison est respectivement de 60-90 %, 40-70 %, 10-20 % et 0 % pour les stades I, II, III et IV.
La tomodensitométrie thoracique a un grand intérêt diagnostique surtout en cas de présentation peu typique. Le tableau typique32 (fig. 2) est celui de l’association d’adénopathies médiastino-hilaires bilatérales et symétriques associées à des micronodules de distribution périlymphatique (les micronodules suivent les trajets lymphatiques et se retrouvent le long des axes péribronchovasculaires, à l’échelle du lobule secondaire dans les septums interlobulaires, en sous-pleural et notamment le long des scissures) prédominant dans les territoires supérieurs et moyens (fig. 3). Ce tableau permet de prédire avec plus de 95 % de sécurité un diagnostic de sarcoïdose chez 80 % des patients.2, 33, 34 Dans les rares cas où des lésions non habituelles prédominent, comme le verre dépoli ou les condensations, la présence d’adénopathies typiques ou de micronodules périlymphatiques même au second plan permet dans la plupart des cas de retenir le diagnostic de sarcoïdose.35, 36 La tomodensitométrie permet, en outre, de renseigner avec plus de précision sur le caractère réversible inflammatoire (micronodules, nodules) ou irréversible des lésions (fibrose). Les lésions de fibrose pulmonaire prédominent dans les territoires supérieurs moyens et périhilaires. Le tableau le plus typique est celui de réticulations avec distorsion péribronchovasculaire prédominant dans les lobes supérieurs dont le volume est réduit. Des adéno- athies calcifiées bilatérales peuvent témoigner de la déclaration ancienne de la maladie.37
Les EFR sont souvent normales ou discrètement anormales lors du diagnostic.18 Le retentissement fonctionnel consiste typiquement en un syndrome restrictif et un trouble de diffusion.2, 18 Le suivi de la capacité vitale forcée (CVF) est l’élément le plus fiable indispensable pour le suivi pulmonaire, avec la radiographie simple du thorax.32 Un trouble ventilatoire obstructif est présent dans 14 à 50 % des cas, et plusieurs mécanismes peuvent en être responsables.38 La DLCO est le plus souvent diminuée en raison d’une atteinte de la membrane alvéolocapillaire (par exemple en cas de fibrose) ou être le reflet d’une cause vasculaire (hypertension pulmonaire). Une mesure des débits inspiratoires est recommandée en cas de suspicion d’atteinte laryngée.
Le test de marche des 6 minutes peut mettre en évidence une réduction du parcours et une désaturation en oxygène et, chez les patients avec une dyspnée persistante sous traitement, ces anomalies conduisent à rechercher une hypertension pulmonaire. Une épreuve d’effort sur tapis ou ergocycle s’avère utile lorsque le mécanisme de la dyspnée n’est pas compris en orientant vers une cause pulmonaire, cardiovasculaire, musculaire spécifique et/ou un déconditionnement.
Certaines formes de sarcoïdose sont qualifiées de « dangereuses » car associées à un risque de mortalité, de morbidité significative, ou la nécessité d’un traitement lourd ou exceptionnel (biomédicament anti-TNFα, transplantation) : c’est le cas notamment de certaines atteintes pulmonaires, cardiaques et neurologiques.
Principales formes sévères pulmonaires
Les principales formes sévères sont la fibrose pulmonaire avec retentissement significatif, l’hypertension pulmonaire, la surinfection par une infection pulmonaire aspergillaire et les atteintes graves des voies aériennes avec trouble ventilatoire obstructif.
Fibrose pulmonaire
La fibrose pulmonaire définie par un stade IV radiographique concerne 5,4 % des patients au début de la maladie et jusqu’à 20 % en cours d’évolution.18 Dans les pays occidentaux, elle est la principale cause de morbidité-mortalité, avec une survie de moins de 80 % à 15 ans. Les causes de décès sont l’insuffisance respiratoire terminale et/ou l’hypertension pulmonaire, puis l’hémoptysie massive secondaire à un aspergillome.31 L’évolution peut être également compliquée d’un pneumothorax ou d’infections mycobactériennes non tuberculeuses et de surinfections bronchiques favorisées par les bronchectasies de traction.31 Plus de la moitié des patients ayant une fibrose pulmonaire gardent une activité résiduelle granulomateuse mise en évidence par la tomodensitométrie couplée à la tomographie à émission de positons (TEP/TDM) et peuvent bénéficier d’un traitement par corticoïdes ou immunosuppresseurs.31Hypertension pulmonaire
Elle touche 1-6 % de l’ensemble des patients, mais elle atteint 74 % des candidats à une transplantation.39 Elle est plus fréquente chez les hommes, et en cas de stade radiologique avancé.2 Elle est suspectée en cas de dyspnée persistante, de souffle holosystolique d’insuffisance tricuspidienne, d’éclat de B2 au foyer pulmonaire, d’insuffisance cardiaque droite, de diminution isolée ou disproportionnée de la DLCO (rapport CVF %Pred/DLCO %Pred > 1,4-1,5), de désaturation au TM6 (saturation < 90 %) et d’un rapport de diamètre artère pulmonaire/aorte supérieur à 1. L’échographie cardiaque est un examen de dépistage, mais seul le cathétérisme cardiaque droit confirme le diagnostic. La sévérité est inversement corrélée à la DLCO.40 Il s’agit le plus souvent d’une hypertension pulmonaire précapillaire, le cathétérisme permettant d’éliminer les hypertensions postcapillaires secondaires à une sarcoïdose du ventricule gauche, avec des mécanismes complexes : destruction du lit capillaire par la fibrose, obstruction vasculaire par des adénopathies ou une médiastinite fibreuse, atteinte vasculaire spécifique granulomateuse.39 L’embolie pulmonaire ou un syndrome d’apnées du sommeil sont à éliminer. L’hypertension pulmonaire a un pronostic sombre, avec une survie de 55 % à 5 ans.29, 31 La prise en charge diagnostique et thérapeutique doit être réalisée dans un centre expert. Les immunosuppresseurs peuvent améliorer l’hémodynamique chez certains patients sélectionnés (adénopathies médiastinales compressives hypermétaboliques au 18FFDG-TEP ou atteinte pulmonaire sans fibrose).29, 41Infections aspergillaires
Elles concernent 3-12 % des patients42 et compliquent le plus souvent des formes pulmonaires avancées, de stade IV chez des patients exposés antérieurement aux moisissures dans leur emploi.43 À la tomodensitométrie, on note une image de grelot déclive ou un épaississement de la paroi d’une cavité. Une sérologie aspergillaire positive et la mise en évidence d’Aspergillus avec fongigramme dans un prélèvement respiratoire sont habituellement positives. Le risque est celui de l’hémoptysie massive, et la difficulté de prise en charge réside dans la gestion des anti-inflammatoires dans ce contexte infectieux, les antifongiques apportant un bénéfice.43Trouble ventilatoire obstructif
Associé à une mortalité accrue,31 il est souvent secondaire à plusieurs mécanismes intriqués, étudiés par la tomodensitométrie thoracique et l’endoscopie bronchique. Les principaux sont l’existence de distorsions bronchiques par la fibrose, un épaississement bronchique par infiltration granulomateuse, parfois sténosante.44 Plus rarement, le trouble ventilatoire obstructif peut résulter d’une compression ganglionnaire extrinsèque, de sténoses significatives sur des gros troncs bronchiques ou d’une bronchiolite.Sarcoïdose cardiaque
La sarcoïdose cardiaque45 est rarement patente (2-5 %), mais représente la deuxième cause de mortalité de sarcoïdose, principalement liée à une insuffisance cardiaque ou à une mort subite.30 Le facteur de plus mauvais pronostic en cas d’atteinte cardiaque est l’extension de la dysfonction du ventricule gauche.45 La survie à 5 ans a été évaluée dans une ancienne étude à 57 % en cas de fraction d’éjection du ventricule gauche (FEVG) inférieure à 30 %.46 L’atteinte cardiaque est caractérisée par une infiltration hétérogène du myocarde par les granulomes avec une prédominance pour la paroi libre du ventricule gauche, le septum et les voies de conduction qui s’y situent. L’atteinte du ventricule droit est moins fréquente mais de moins bon pronostic car témoignant probablement d’une extension plus importante à partir du ventricule gauche. Devant tout cas de sarcoïdose et à chaque consultation lors du suivi, il faut rechercher des signes fonctionnels cardiaques : douleurs thoraciques, palpitations soutenues, dyspnée, syncope. L’atteinte cardiaque peut se révéler par des troubles de la conduction (notamment bloc auriculo-ventriculaire, [BAV] de divers degrés et bloc de branches surtout droit), des troubles du rythme (auriculaires mais surtout tachycardie ventriculaire) une insuffisance cardiaque ou une mort subite.45 L’ECG est indispensable au diagnostic de n’importe quelle forme de sarcoïdose et à renouveler périodiquement (au moins une fois par an). Une échographie cardiaque transthoracique est réalisée quasi constamment en pratique, avec recherche d’une dysfonction ventriculaire gauche en premier lieu, d’une dysfonction/dilatation du ventricule droit, d’un amincissement du septum basal, d’anomalies de la cinétique, d’anomalies de la paroi du ventricule comme un anévrisme. Les dosages de la troponine et de la fraction terminale du peptide natriurétique de type B (NT-proBNP) ne sont ni sensibles ni spécifiques mais peuvent être utilisés devant des signes cliniques évocateurs d’une atteinte cardiaque. Le holter ECG est réalisé en cas de palpitations, d’anomalie de l’ECG et de doute clinique d’atteinte cardiaque.
L’imagerie par résonance magnétique (IRM) cardiaque et la TEP/TDM au 18FFDG (examens à réaliser dans des centres experts) confirment ou non l’atteinte cardiaque et la présence de lésions actives, idéalement lors de réunions multidisciplinaires avec un cardiologue, un expert en sarcoïdose et des spécialistes dédiés en imagerie cardiologique. Plusieurs consensus ont été établis pour le diagnostic de sarcoïdose cardiaque.47-49 Le consensus international de la Hearth Rythm Society (HRS) distingue deux tableaux :
– l’un très rare avec des granulomes myocardiques après élimination de tout autre diagnostic ;
– le second défini par 3 critères : présence de granulomes dans un autre organe ; exclusion des autres causes pouvant expliquer les manifestations cardiaques (cardiopathie ischémique, remodelage cardiaque lié à une hypertension pulmonaire…) ; et au moins une manifestation parmi les suivantes : cardiomyopathie ou troubles de la conduction résolutifs sous corticoïdes et/ou immunosuppresseurs, BAV de haut degré (BAV II de type Mobitz II ; BAV III), tachycardie ventriculaire soutenue, FEVG < 40 % en échographie ; rehaussement tardif sous gadolinium en IRM ; fixation hétérogène en 18FFDG-TEP.
La prise en charge est multidisciplinaire et discute du traitement immunosuppresseur, d’un traitement médicamenteux à visée cardiaque (bêtabloquant, inhibiteur de l’enzyme de conversion, antiarythmique) et de la mise en place d’un défibrillateur implantable ou d’un entraînement électrosystolique intracardiaque.45
L’imagerie par résonance magnétique (IRM) cardiaque et la TEP/TDM au 18FFDG (examens à réaliser dans des centres experts) confirment ou non l’atteinte cardiaque et la présence de lésions actives, idéalement lors de réunions multidisciplinaires avec un cardiologue, un expert en sarcoïdose et des spécialistes dédiés en imagerie cardiologique. Plusieurs consensus ont été établis pour le diagnostic de sarcoïdose cardiaque.47-49 Le consensus international de la Hearth Rythm Society (HRS) distingue deux tableaux :
– l’un très rare avec des granulomes myocardiques après élimination de tout autre diagnostic ;
– le second défini par 3 critères : présence de granulomes dans un autre organe ; exclusion des autres causes pouvant expliquer les manifestations cardiaques (cardiopathie ischémique, remodelage cardiaque lié à une hypertension pulmonaire…) ; et au moins une manifestation parmi les suivantes : cardiomyopathie ou troubles de la conduction résolutifs sous corticoïdes et/ou immunosuppresseurs, BAV de haut degré (BAV II de type Mobitz II ; BAV III), tachycardie ventriculaire soutenue, FEVG < 40 % en échographie ; rehaussement tardif sous gadolinium en IRM ; fixation hétérogène en 18FFDG-TEP.
La prise en charge est multidisciplinaire et discute du traitement immunosuppresseur, d’un traitement médicamenteux à visée cardiaque (bêtabloquant, inhibiteur de l’enzyme de conversion, antiarythmique) et de la mise en place d’un défibrillateur implantable ou d’un entraînement électrosystolique intracardiaque.45
Neurosarcoïdose
La neurosarcoïdose est rare, touchant 5 à 10 % des patients et peut atteindre n’importe quelle partie du système nerveux central (85 % des cas) ou périphérique. En cas de neurosarcoïdose, les symptômes sont révélateurs de la sarcoïdose dans plus de la moitié des cas.50
Les atteintes préférentielles du système nerveux central sont : méningées, médullaires, intraparenychymateuses cérébrales, des nerfs crâniens : V, VII, nerf optique, médullaire (troubles moteurs sensitifs ou sphinctérieux), hypothalamo- hypophysaires (atteinte hypophyse antérieure avec insuffisance gonadique pouvant aller jusqu’au pan- hypopituitarisme et/ou diabète insipide). Des troubles cognitifs sont présents chez 20 % des patients, en particulier en cas d’atteinte méningée. Les crises convulsives sont possibles, les atteintes vasculaires cérébrales sont des manifestations rares mais associées à un risque de décès important (> 20 %).51
Le système nerveux périphérique est atteint dans 15 % des cas de neurosarcoïdose. Les localisations neurologiques sont graves, avec dans deux tiers des cas des séquelles. Le pronostic est moins bon en en cas d’atteinte médullaire et du système nerveux périphérique. Les principaux diagnostics différentiels à éliminer sont les causes infectieuses (tuberculose, leuco-encéphalopathie multifocale progressive [LEMP], virales, maladie de Lyme, syphilis), cancer, lymphomes, sclérose en plaques. L’analyse du liquide céphalorachidien (LCR) met en évidence une méningite lymphocytaire avec parfois une hypoglycorachie. L’IRM cérébrale et médullaire peut montrer, en cas d’atteinte évocatrice, une prise de contraste leptoméningée avec un aspect micronodulaire confluent.
Un consensus récent a établi des critères diagnostiques.52 Le diagnostic de neurosarcoïdose est retenu en cas de la présence des deux critères :
– présentation clinique et évaluation diagnostique suggérant une neurosarcoïdose, définie par le tableau clinique, l’IRM, l’analyse du LCR et/ou l’électromyogramme avec des données typiques d’une inflammation granulomateuse et l’exclusion rigoureuse d’autres causes ;
– atteinte histologique extraneurologique (diagnostic probable) et/ou atteinte histologique neurologique (diagnostic certain).
Le diagnostic est considéré comme possible lorsque le tableau est compatible (critère 1) mais sans confirmation histologique. L’évolution sous traitement ne fait pas partie des critères, contrairement à la sarcoïdose cardiaque, car certaines infections, cancers, maladies inflammatoires peuvent répondre initialement au traitement anti-inflammatoire.
Il n’existe pas d’essai randomisé actuellement disponible dans les atteintes neurologiques, le traitement chez ces patients doit être prolongé, avec un avis neurologique spécialisé.
Les atteintes préférentielles du système nerveux central sont : méningées, médullaires, intraparenychymateuses cérébrales, des nerfs crâniens : V, VII, nerf optique, médullaire (troubles moteurs sensitifs ou sphinctérieux), hypothalamo- hypophysaires (atteinte hypophyse antérieure avec insuffisance gonadique pouvant aller jusqu’au pan- hypopituitarisme et/ou diabète insipide). Des troubles cognitifs sont présents chez 20 % des patients, en particulier en cas d’atteinte méningée. Les crises convulsives sont possibles, les atteintes vasculaires cérébrales sont des manifestations rares mais associées à un risque de décès important (> 20 %).51
Le système nerveux périphérique est atteint dans 15 % des cas de neurosarcoïdose. Les localisations neurologiques sont graves, avec dans deux tiers des cas des séquelles. Le pronostic est moins bon en en cas d’atteinte médullaire et du système nerveux périphérique. Les principaux diagnostics différentiels à éliminer sont les causes infectieuses (tuberculose, leuco-encéphalopathie multifocale progressive [LEMP], virales, maladie de Lyme, syphilis), cancer, lymphomes, sclérose en plaques. L’analyse du liquide céphalorachidien (LCR) met en évidence une méningite lymphocytaire avec parfois une hypoglycorachie. L’IRM cérébrale et médullaire peut montrer, en cas d’atteinte évocatrice, une prise de contraste leptoméningée avec un aspect micronodulaire confluent.
Un consensus récent a établi des critères diagnostiques.52 Le diagnostic de neurosarcoïdose est retenu en cas de la présence des deux critères :
– présentation clinique et évaluation diagnostique suggérant une neurosarcoïdose, définie par le tableau clinique, l’IRM, l’analyse du LCR et/ou l’électromyogramme avec des données typiques d’une inflammation granulomateuse et l’exclusion rigoureuse d’autres causes ;
– atteinte histologique extraneurologique (diagnostic probable) et/ou atteinte histologique neurologique (diagnostic certain).
Le diagnostic est considéré comme possible lorsque le tableau est compatible (critère 1) mais sans confirmation histologique. L’évolution sous traitement ne fait pas partie des critères, contrairement à la sarcoïdose cardiaque, car certaines infections, cancers, maladies inflammatoires peuvent répondre initialement au traitement anti-inflammatoire.
Il n’existe pas d’essai randomisé actuellement disponible dans les atteintes neurologiques, le traitement chez ces patients doit être prolongé, avec un avis neurologique spécialisé.
Autres atteintes d’organes
Atteinte cutanée
C’est la plus fréquente des atteintes extrathoraciques (> 30 %).53, 54 Elle peut souvent être révélatrice de la maladie et est plus fréquente chez les femmes. En dehors du syndrome de Löfgren, les plaques et les papules (petits et gros nodules) sont les lésions les plus fréquentes (fig. 4 et 5). Ces lésions sont fermes, non inflammatoires sans topographie particulière, mais l’atteinte faciale, des cicatrices ou tatouage est classique.55 Le lupus pernio (fig. 6), de plus mauvais pronostic, se présente comme une infiltration violacée du nez avec extension possible aux joues et aux lobes des oreilles ; il est souvent associé à une atteinte ORL.
Atteintes ophtalmologiques
Elles sont rapportées chez 15 à 30 % des patients.56 Un consensus international a défini les différents critères diagnostiques d’une sarcoïdose ophtalmique allant de diagnostic défini à possible.57 L’uvéite est l’atteinte la plus fréquente (30-70 %), avec en premier lieu l’uvéite antérieure, bilatérale dans trois quarts des cas. La clinique est caractérisée par un œil rouge douloureux et une photophobie, mais dans un tiers des cas le patient est asymptomatique, justifiant un examen ophtalmologique spécialisé systématique.58 L’atteinte postérieure peut se compliquer d’œdème maculaire, de néovascularisation choroïdienne ou de glaucome. Les autres atteintes, par ordre de fréquence décroissant, sont la kératite sèche due à l’atteinte des glandes lacrymales, et l’atteinte des conjonctives (conjonctivite ou granulome). L’atteinte des annexes (glandes lacrymales, paupières) représente 10 % des atteintes ophtalmiques. La neuropathie optique présente dans 5 % des cas est la plus fréquente des atteintes neuro-ophtalmologiques. L’atteinte ophtalmique est généralement de bon pronostic, à condition que la prise en charge soit précoce. Une récente étude montrait que seulement 2,4 % des patients souffrant d’uvéite gardaient des séquelles, avec une baisse de l’acuité visuelle sévère.59
Atteintes hépatiques
Les atteintes hépatiques (15 %) se caractérisent par des anomalies du bilan hépatique, le plus souvent à type de cholestase anictérique et par l’absence de symptômes. Les douleurs abdominales et l’hépatomégalie sont néanmoins possibles. La cirrhose et l’hypertension portales sont rares.60
Atteintes oto-rhino-laryngées
L’atteinte ORL (2-4 %) peut se traduire par une rhinite croûteuse, une parotidite, ou une atteinte laryngée particulièrement sévère.61, 62
Atteinte rénale
L’atteinte rénale (2 %) consiste typiquement en une néphropathie tubulo-interstitielle granulomateuse, parfois avec néphrocalcinose qui peut entraîner une insuffisance rénale aiguë et conduire parfois à une dialyse.63
Atteinte musculaire
Rare, elle peut se présenter sous différentes formes : nodulaire, indolente, myopathique ou neuromyopathique de moins bon pronostic.64 Les autres atteintes rares sont osseuses, digestives, génito-urinaires.
Signes d’inconfort persistants ou syndrome parasarcoïdien
Le terme « syndrome parasarcoïdien » a été employé pour désigner les manifestations accompagnant la maladie mais non causées directement par les granulomes.65 Ces manifestations sont parfois extrêmement invalidantes et comprennent : la neuropathie des petites fibres66, 67 caractérisée par l’association de douleurs neuropathiques et une atteinte du système nerveux autonome (palpitations, hypotension orthostatique, dyshidrose, troubles du transit) ; les troubles cognitifs, la dépression et des syndromes algiques articulaires et musculaires en dehors d’anomalies objectives ; la fatigue touchant 50 à 70 % des patients, plus particulièrement les femmes,20 qui peut être profonde et compromettre la vie socioprofessionnelle et personnelle. Elle peut résulter de l’activité de la maladie, d’une corticothérapie prolongée, d’un trouble métabolique (diabète, insuffisance surrénalienne, hypothyroïdie), ou d’un syndrome d’apnées obstructives du sommeil plus fréquent dans cette population ou enfin ne pas avoir de mécanisme individualisable.
Évolution
On différencie les formes d’évolution courte des formes persistantes selon qu’elles durent moins ou plus de 5 ans. Dans la moitié des cas, la sarcoïdose est spontanément résolutive 2 ans après le diagnostic.3 Certains paramètres sont prédictifs3 d’une résolution rapide : un syndrome de Löfgren, une uvéite aiguë, un stade I, et certains haplotypes (HLA-DRB1*03). D’autres prédisent une évolution chronique : un âge de début inférieur à 40 ans, une origine afro-caribéenne, certaines atteintes extrathoraciques (cardiaque, neurologique, rénale, ORL, lupus pernio, uvéite postérieure, néphro- calcinose).
Morbidité et mortalité
Le risque relatif de maladie thromboembolique, de lymphoprolifération et de pathologie auto-immune (syndrome de Sjögren, spondylarthrite ankylosante) est augmenté dans la sarcoïdose.1, 3, 68 Le syndrome d’apnées obstructives du sommeil est également accru mais d’origine multifactorielle.69
Le pronostic de la sarcoïdose est favorable dans 80 % des cas, avec ou sans traitement ; 10-20 % des patients vont garder des séquelles et 1-5 % des patients vont mourir de leur sarcoïdose. La première cause de décès de sarcoïdose est liée aux atteintes pulmonaires sévères suivies par les atteintes cardiaques.30, 70, 71 Globalement, la sarcoïdose entraîne une diminution de l’espérance de vie.30 Une étude suédoise a montré le contraste entre patients ne nécessitant pas de traitement pour lesquels il n’y a pas de surmortalité et les patients ayant une nécessité d’être traités pour lesquels la mortalité est augmentée.72 Dans les formes pulmonaires, l’extension de la fibrose supérieure à 20 % à la tomodensitométrie, et la présence d’une hypertension pulmonaire estimée sur l’augmentation du rapport du diamètre artère pulmonaire (AP)/aorte (Ao) supérieur à 1 à la tomodensitométrie injectée ou par un cathétérisme cardiaque sont des facteurs de risque reconnus de surmortalité.73, 74
Le pronostic de la sarcoïdose est favorable dans 80 % des cas, avec ou sans traitement ; 10-20 % des patients vont garder des séquelles et 1-5 % des patients vont mourir de leur sarcoïdose. La première cause de décès de sarcoïdose est liée aux atteintes pulmonaires sévères suivies par les atteintes cardiaques.30, 70, 71 Globalement, la sarcoïdose entraîne une diminution de l’espérance de vie.30 Une étude suédoise a montré le contraste entre patients ne nécessitant pas de traitement pour lesquels il n’y a pas de surmortalité et les patients ayant une nécessité d’être traités pour lesquels la mortalité est augmentée.72 Dans les formes pulmonaires, l’extension de la fibrose supérieure à 20 % à la tomodensitométrie, et la présence d’une hypertension pulmonaire estimée sur l’augmentation du rapport du diamètre artère pulmonaire (AP)/aorte (Ao) supérieur à 1 à la tomodensitométrie injectée ou par un cathétérisme cardiaque sont des facteurs de risque reconnus de surmortalité.73, 74
Traitement et suivi
Principes du traitement
La mise en œuvre d’un traitement peut ne pas être nécessaire s’il n’y a pas d’atteinte sévère ou de syndrome parasarcoïdien, ou être nécessaire d’emblée ou de façon différée devant une progression secondaire. Le traitement de la sarcoïdose, qui repose sur des médicaments anti-inflammatoires (corticostéroïdes, immunosuppresseurs, anti-TNFα), n’est pas curatif, mais suspensif sur les manifestations en contrôlant la réaction granulomateuse. Le traitement n’a aucun effet sur la fibrose une fois installée. La corticothérapie est le traitement de première ligne. Il n’y a pas de consensus sur la dose initiale (entre 1/3 et 1 mg/kg/j équivalent prednisone), la durée du traitement (le plus souvent 12 mois ou plus) et les modalités de décroissance. La décroissance thérapeutique doit être progressive, avec parfois la mise en évidence d’une dose seuil qui permet de contrôler la maladie. Des rechutes sont fréquentes lors de la diminution des doses, et surtout après l’arrêt du traitement. En dehors de la corticothérapie, les autres molécules disponibles ont un délai d’action retardé de plusieurs mois. Chaque traitement doit être encadré par une prévention de ses effets indésirables. Des traitements symptomatiques ou dirigés sur certains organes peuvent aussi être nécessaires et compléter le traitement de la sarcoïdose.
Indications
Le traitement n’est pas systématique, et la décision de traiter nécessite de répondre à certaines questions au préalable : le patient est-il symptomatique ? les symptômes peuvent-ils être contrôlés par une thérapeutique locale ? existe-t-il une atteinte d’organes vitaux ? le patient a-t-il une forme chronique de la maladie ? a-t-il des contre-indications au traitement ? Les principales indications thérapeutiques à visée pulmonaire et extrapulmonaires sont résumées dans les tableaux 3 et 4.
Modalités du traitement
Corticostéroïdes
La corticothérapie est le traitement général de référence. Elle est en règle générale prolongée sur 12 mois ou plus. Des doses de prednisone orale (0,5 mg/kg) sont prescrites à l’initiation du traitement. Deux études récentes ont montré qu’en cas de sarcoïdose pulmonaire récente l’efficacité était obtenue en 1 mois ou moins, et qu’une décroissance rapide des corticoïdes (objectif de dose de 10 mg/j à 3-4 mois) avait une efficacité satisfaisante moyennant une prise de poids moindre qu’une décroissance plus lente.75, 76Les traitements locaux doivent être privilégiés dans les atteintes cutanées (dermocorticoïdes) et l’uvéite antérieure (corticoïdes locaux, injections périoculaires de corticoïdes). La corticothérapie inhalée est discutée dans les formes pulmonaires ne relevant pas d’une corticothérapie systémique, quand la toux est présente, sèche et gênante.
Autres traitements systémiques
D’autres molécules peuvent être prescrites,77 en cas de contre-indication, échec ou mauvaise tolérance des corticoïdes ou à titre d’épargne cortisonique lorsque la dose seuil dépasse durablement 10 mg/j :– les antipaludéens de synthèse (hydroxychloroquine) peuvent être utilisés pour les atteintes cutanées modérées et en cas d’hypercalcémie ; une surveillance ophtalmologique est nécessaire (recherche d’une rétinopathie) ;
– le méthotrexate à faible posologie hebdomadaire est l’immunosuppresseur le plus choisi en deuxième ligne ;78 il est associé à une contraception et à une supplémentation folinique ; ses complications sont d’ordre hématologique, hépatique, pulmonaire et rénal ;
– l’azathioprine, autre molécule de deuxième ligne, est contre-indiquée en cas de prise d’allopurinol ;
– dans certaines formes graves résistant à ces traitements, les anti-TNFα (infliximab voire adalimumab) peuvent être envisagés.
Traitements d’organe
En cas d’atteinte respiratoire peuvent être prescrits selon la nécessité : une oxygénothérapie, le traitement d’infections pulmonaires surajoutées, notamment aspergillaires, et de leurs complications, des sténoses bronchiques par dilatations et stents. Une réhabilitation peut être particulièrement intéressante, avec une diminution de la fatigue.79 Les hémorragies, liées à une infection aspergillaire ou non, peuvent bénéficier de la radiologie interventionnelle.La greffe d’organe peut être envisagée en cas de stade terminal de la maladie après avoir épuisé toutes les options médicamenteuses ; la survie est identique aux autres indications dans une étude portant sur 700 patients atteints de sarcoïdose transplantés pulmonaires.80
Les traitements des atteintes extra-thoraciques sont variés : traitement médicamenteux de l’insuffisance cardiaque et antiarythmiques, défibrillateur ou entraînement électro-systolique (atteinte cardiaque), anti- épileptiques, dérivation ventriculaire (atteinte neurologique), dialyse, mesures hygiéno-diététiques pour l’hypercalcémie…
Suivi
Les patients doivent être suivis tous les 3 à 6 mois jusqu’à ce que la guérison soit assurée. Chaque visite doit comprendre une recherche d’effets indésirables liés aux traitements, un examen physique complet, une biologie standard et un ECG au moins annuel. Étant donné sa quasi-constance, l’atteinte respiratoire doit être systématiquement surveillée. Plus que le suivi radiographique, toujours recommandé, l’étude de la fonction respiratoire (surtout la capacité vitale forcée) est l’examen le plus intéressant.81 Les EFR doivent être répétées au moins tous les 6 mois (souvent spirométrie tous les 3 mois et transfert pulmonaire du monoxyde de carbone [TLCO] tous les 6 mois). Une dyspnée persistante mal expliquée doit faire rechercher une hypertension pulmonaire ou cardiaque. Elles constituent le paramètre de suivi le plus fidèle pour dépister une aggravation ou pour apprécier la réponse à un éventuel traitement.81 Au stade de fibrose, les poussées résultent pour moitié d’une exacerbation sarcoïdosique et pour l’autre moitié de complications diverses. Celles-ci sont surtout infectieuses, favorisées par les bronchectasies ou distorsions bronchiques. Une surveillance biologique comportant un hémogramme, une créatininémie et une calcémie est justifiée périodiquement. Une atteinte extrapulmonaire (œil, cœur, système nerveux, etc.) nécessite un suivi adapté. Les signes d’inconfort persistants nécessitent une bonne évaluation et une prise en charge adaptée.
Les manifestations de la sarcoïdose, le traitement et ses objectifs doivent être clairement expliqués aux patients.
Les rechutes surviennent le plus souvent dans les 2-6 mois qui suivent l’interruption du traitement et sont exceptionnelles après 3 ans de recul sans traitement. La guérison de la maladie est définie par une rémission stable en dehors de tout traitement pendant 36 mois.
Les manifestations de la sarcoïdose, le traitement et ses objectifs doivent être clairement expliqués aux patients.
Les rechutes surviennent le plus souvent dans les 2-6 mois qui suivent l’interruption du traitement et sont exceptionnelles après 3 ans de recul sans traitement. La guérison de la maladie est définie par une rémission stable en dehors de tout traitement pendant 36 mois.
Références
1. Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 1999;160:736‑55.
2. Spagnolo P, Rossi G, Trisolini R, Sverzellati N, Baughman RP, Wells AU. Pulmonary sarcoidosis. Lancet Respir Med 2018;6:389-402.
3. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014;383:1155‑67.
4. Duchemann B, Annesi-Maesano I, Jacobe de Naurois C, et al. Prevalence and incidence of interstitial lung diseases in a multi- ethnic county of Greater Paris. Eur Respir J 2017;50(2).
5. Arkema EV, Grunewald J, Kullberg S, Eklund A, Askling J. Sarcoidosis incidence and prevalence: a nationwide register- based assessment in Sweden. Eur Respir J 2016;48:1690‑9.
6. Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc 2016;13:1244‑52.
7. Yoon HY, Kim HM, Kim YJ, Song JW. Prevalence and incidence of sarcoidosis in Korea: a nationwide population-based study. Respir Res 2018;19:158.
8. Cozier YC, Govender P, Berman JS. Obesity and sarcoidosis: consequence or contributor? Curr Opin Pulm Med 2018;24:487‑94.
9. Rossides M, Grunewald J, Eklund A, et al. Familial aggregation and heritability of sarcoidosis: a Swedish nested case- control study. Eur Respir J 2018;52(2).
10. Broos CE, van Nimwegen M, Hoogsteden HC, Hendriks RW, Kool M, van den Blink B. Granuloma formation in pulmonary sarcoidosis. Front Immunol 2013;4:437.
11. Ungprasert P, Crowson CS, Matteson EL. Seasonal variation in incidence of sarcoidosis: a population-based study 1976–2013. Thorax 2016;71:1164‑6.
12. Hena KM, Yip J, Jaber N, et al. Clinical course of sarcoidosis in world trade center-exposed firefighters. Chest 2018;153:114‑23.
13. Esteves T, Aparicio G, Garcia-Patos V. Is there any association between Sarcoidosis and infectious agents?: a systematic review and meta-analysis. BMC Pulm Med 2016;16:165.
14. Beghè D, Dall’Asta L, Garavelli C, et al. Sarcoidosis in an Italian province. Prevalence and environmental risk factors. PloS One 2017;12:e0176859.
15. Chen ES. Innate immunity in sarcoidosis pathobiology. Curr Opin Pulm Med 2016;22:469‑75.
16. Judson MA, Thompson BW, Rabin DL, et al. The diagnostic pathway to sarcoidosis. Chest 2003;123:406‑12.
17. Mañá J, Rubio-Rivas M, Villalba N, et al. Multidisciplinary approach and long-term follow-up in a series of 640 consecutive patients with sarcoidosis: Cohort study of a 40-year clinical experience at a tertiary referral center in Barcelona, Spain. Medicine (Baltimore) 2017;96:e7595.
18. Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001;164(10 Pt 1):1885‑9.
19. Grunewald J, Eklund A. Sex-specific manifestations of Löfgren’s syndrome. Am J Respir Crit Care Med 2007;175:40‑4.
20. Drent M, Lower EE, De Vries J. Sarcoidosis-associated fatigue. Eur Respir J 2012;40:255‑63.
21. Bickett AN, Lower EE, Baughman RP. Sarcoidosis diagnostic score: a systematic evaluation to enhance the diagnosis of sarcoidosis. Chest 2018;154:1052-60.
22. Boussouar S, Medjhoul A, Bernaudin JF, et al. Diagnostic efficacy of ultrasound-guided core-needle biopsy of peripheral lymph nodes in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2015;32:188‑93.
23. Sehgal IS, Dhooria S, Aggarwal AN, Agarwal R. Impact of rapid on-site cytological evaluation (rose) on the diagnostic yield of transbronchial needle aspiration during mediastinal lymph node sampling: systematic review and meta-analysis. Chest 2017;153:929-38.
24. Pajares V, Puzo C, Castillo D, et al. Diagnostic yield of transbronchial cryobiopsy in interstitial lung disease: a randomized trial. Respirol Carlton Vic 2014;19:900‑6.
25. Hendriks C, Drent M, Elfferich M, De Vries J. The Fatigue Assessment Scale: quality and availability in sarcoidosis and other diseases. Curr Opin Pulm Med 2018;24:495‑503.
26. Schupp JC, Freitag-Wolf S, Bargagli E, et al. Phenotypes of organ involvement in sarcoidosis. Eur Respir J 2018;51(1).
27. Mahévas M, Chiche L, Uzunhan Y, et al. Association of sarcoidosis and immune thrombocytopenia: presentation and outcome in a series of 20 patients. Medicine (Baltimore) 2011;90:269‑78.
28. Piekarski E, Benali K, Rouzet F. Nuclear imaging in sarcoidosis. Semin Nucl Med 2018;48:246‑60.
29. Boucly A, Cottin V, Nunes H, et al. Management and long-term outcomes of sarcoidosis-associated pulmonary hypertension. Eur Respir J 2017;50(4).
30. Jamilloux Y, Maucort-Boulch D, Kerever S, et al. Sarcoidosis-related mortality in France: a multiple-cause-of-death analysis. Eur Respir J 2016;48:1700‑9.
31. Nardi A, Brillet PY, Letoumelin P, et al. Stage IV sarcoidosis: comparison of survival with the general population and causes of death. Eur Respir J 2011;38:1368‑73.
32. Nunes H, Uzunhan Y, Gille T, Lamberto C, Valeyre D, Brillet PY. Imaging of sarcoidosis of the airways and lung parenchyma and correlation with lung function. Eur Respir J 2012;40:750‑65.
33. Grenier P, Chevret S, Beigelman C, Brauner MW, Chastang C, Valeyre D. Chronic diffuse infiltrative lung disease: determination of the diagnostic value of clinical data, chest radiography, and CT and Bayesian analysis. Radiology 1994;191:383‑90.
34. Müller NL, Kullnig P, Miller RR. The CT findings of pulmonary sarcoidosis: analysis of 25 patients. Am J Roentgenol 1989;152:1179‑82.
35. Martin SG, Kronek LP, Valeyre D, et al. High-resolution computed tomography to differentiate chronic diffuse interstitial lung diseases with predominant ground-glass pattern using logical analysis of data. Eur Radiol 2010;20:1297‑310.
36. De Margerie-Mellon C, Dion G, Darlay J, et al. High-resolution computed tomography to differentiate chronic diffuse infiltrative lung diseases with chronic multifocal consolidation patterns using logical analysis of data. Sarcoidosis Vasc Diffuse Lung Dis 2016;33:355‑71.
37. Abehsera M, Valeyre D, Grenier P, Jaillet H, Battesti JP, Brauner MW. Sarcoidosis with pulmonary fibrosis: CT patterns and correlation with pulmonary function. Am J Roentgenol 2000;174:1751‑7.
38. Mihailovic-Vucinic V, Jovanovic D. Pulmonary sarcoidosis. Clin Chest Med 2008;29:459‑73.
39. Nunes H, Uzunhan Y, Freynet O, et al. Pulmonary hypertension complicating sarcoidosis. Presse Med 2012;41:e303‑16.
40. Baughman RP, Shlobin OA, Wells AU, et al. Clinical features of sarcoidosis associated pulmonary hypertension: Results of a multi-national registry. Respir Med 2018;139:72‑8.
41. Baughman RP, Culver DA, Cordova FC, et al. Bosentan for sarcoidosis-associated pulmonary hypertension: a double-blind placebo controlled randomized trial. Chest 2014;145:810‑7.
42. Denning DW, Pleuvry A, Cole DC. Global burden of chronic pulmonary aspergillosis complicating sarcoidosis. Eur Respir J 2013;41:621‑6.
43. Uzunhan Y, Nunes H, Jeny F, et al. Chronic pulmonary aspergillosis complicating sarcoidosis. Eur Respir J 2017;49(6).
44. Naccache JM, Lavolé A, Nunes H, et al. High-resolution computed tomographic imaging of airways in sarcoidosis patients with airflow obstruction. J Comput Assist Tomogr 2008;32:905‑12.
45. Birnie DH, Sauer WH, Judson MA. Consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Br Card Soc 2016;102:411‑4.
46. Chiu CZ, Nakatani S, Zhang G, et al. Prevention of left ventricular remodeling by long-term corticosteroid therapy in patients with cardiac sarcoidosis. Am J Cardiol 2005;95:143‑6.
47. Judson MA, Costabel U, Drent M, et al. The WASOG Sarcoidosis Organ Assessment Instrument: An update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:19‑27.
48. Terasaki F, Yoshinaga K. New guidelines for diagnosis of cardiac sarcoidosis in Japan. Ann Nucl Cardiol 2017;3:42‑5.
49. Birnie DH, Sauer WH, Bogun F, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm 2014;11:1305‑23.
50. Cohen Aubart F, Galanaud D, Haroche J, et al. Les atteintes neurologiques au cours de la sarcoïdose : diagnostic et traitement. Rev Med Interne 2017;38:393‑401.
51. Jachiet V, Lhote R, Rufat P, et al. Clinical, imaging, and histological presentations and outcomes of stroke related to sarcoidosis. J Neurol 2018;256:2333-41.
52. Stern BJ, Royal W, Gelfand JM, et al. Definition and consensus diagnostic criteria for neurosarcoidosis: from the Neurosarcoidosis Consortium Consensus Group. JAMA Neurol 2018;75:1546-53.
53. Ungprasert P, Wetter DA, Crowson CS, Matteson EL. Epidemiology of cutaneous sarcoidosis, 1976-2013: a population-based study from Olmsted County, Minnesota. J Eur Acad Dermatol Venereol 2016;30:1799‑804.
54. Noe MH, Rosenbach M. Cutaneous sarcoidosis. Curr Opin Pulm Med 2017;23:482‑6.
55. Kluger N. Sarcoidosis on tattoos: a review of the literature from 1939 to 2011. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:86‑102.
56. Sève P, Kodjikian L, Jamilloux Y. Manifestations ophtalmologiques de la sarcoidose : que doit savoir l’interniste ? Rev Med Interne 2018;39:728‑37.
57. Herbort CP, Rao NA, Mochizuki M, members of Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop On Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm 2009;17:160‑9.
58. Rothova A, Alberts C, Glasius E, Kijlstra A, Buitenhuis HJ, Breebaart AC. Risk factors for ocular sarcoidosis. Doc Ophthalmol Adv Ophthalmol 1989;72:287‑96.
59. Rochepeau C, Jamilloux Y, Kerever S, et al. Long-term visual and systemic prognoses of 83 cases of biopsy- proven sarcoid uveitis. Br J Ophthalmol 2017;101:856‑61.
60. Deutsch-Link S, Fortuna D, Weinberg EM. A comprehensive review of hepatic sarcoid. Semin Liver Dis 2018;38:284‑97.
61. Aubart FC, Ouayoun M, Brauner M, et al. Sinonasal involvement in sarcoidosis: a case-control study of 20 patients. Medicine (Baltimore) 2006;85:365‑71.
62. Duchemann B, Lavolé A, Naccache JM, et al. Laryngeal sarcoidosis: a case- control study. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:227‑34.
63. Bergner R, Hoffmann M, Waldherr R, Uppenkamp M. Frequency of kidney disease in chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2003;20:126‑32.
64. Cohen Aubart F, Abbara S, Maisonobe T, Cottin V, Papo T, Haroche J, et al. Symptomatic muscular sarcoidosis: Lessons from a nationwide multicenter study. Neurol Neuroimmunol Neuroinflammation 2018;5:e452.
65. Judson MA. The clinical features of sarcoidosis: a comprehensive review. Clin Rev Allergy Immunol 2015;49:63‑78.
66. Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: Clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med 2017;126:135‑8.
67. Heij L, Niesters M, Swartjes M, et al. Safety and efficacy of ARA 290 in sarcoidosis patients with symptoms of small fiber neuropathy: a randomized, double-blind pilot study. Mol Med Camb Mass 2012;18:1430‑6.
68. Wu CH, Chung PI, Wu CY, et al. Comorbid autoimmune diseases in patients with sarcoidosis: A nationwide case-control study in Taiwan. J Dermatol 2017;44:423‑30.
69. Bosse-Henck A, Koch R, Wirtz H, Hinz A. Fatigue and excessive daytime sleepiness in sarcoidosis: prevalence, predictors, and relationships between the two symptoms. Respir Int Rev Thorac Dis 2017;94:186‑97.
70. Reich JM. Mortality of intrathoracic sarcoidosis in referral vs population- based settings*: influence of stage, ethnicity, and corticosteroid therapy. CHEST J 2002;121:32.
71. Gribbin J, Hubbard RB, Le Jeune I, Smith CJP, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006;61:980‑5.
72. Rossides M, Kullberg S, Askling J, Eklund A, Grunewald J, Arkema EV. Sarcoidosis mortality in Sweden: a population-based cohort study. Eur Respir J 2018;51(2).
73. Walsh SL, Wells AU, Sverzellati N, et al. An integrated clinicoradiological staging system for pulmonary sarcoidosis: a case-cohort study. Lancet Respir Med 2014;2:123‑30.
74. Kirkil G, Lower EE, Baughman RP. Predictors of mortality in pulmonary sarcoidosis. Chest 2018;153:105‑13.
75. Broos CE, Poell LHC, Looman CWN, et al. No evidence found for an association between prednisone dose and FVC change in newly-treated pulmonary sarcoidosis. Respir Med 2018;138S:S31‑7.
76. Broos CE, Wapenaar M, Looman CWN, et al. Daily home spirometry to detect early steroid treatment effects in newly treated pulmonary sarcoidosis. Eur Respir J 2018;51(1).
77. James WE, Baughman R. Treatment of sarcoidosis: grading the evidence. Expert Rev Clin Pharmacol 2018;11:677‑87.
78. Schutt AC, Bullington WM, Judson MA. Pharmacotherapy for pulmonary sarcoidosis: a Delphi consensus study. Respir Med 2010;104:717‑23.
79. Strookappe B, Swigris J, De Vries J, Elfferich M, Knevel T, Drent M. Benefits of physical training in sarcoidosis. Lung 2015;193:701‑8.
80. Taimeh Z, Hertz MI, Shumway S, Pritzker M. Lung transplantation for pulmonary sarcoidosis. Twenty-five years of experience in the USA. Thorax 2016;71:378‑9.
81. Zappala CJ, Desai SR, Copley SJ, et al. Accuracy of individual variables in the monitoring of long-term change in pulmonary sarcoidosis as judged by serial high-resolution CT scan data. Chest 2014;145:101‑7.
2. Spagnolo P, Rossi G, Trisolini R, Sverzellati N, Baughman RP, Wells AU. Pulmonary sarcoidosis. Lancet Respir Med 2018;6:389-402.
3. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014;383:1155‑67.
4. Duchemann B, Annesi-Maesano I, Jacobe de Naurois C, et al. Prevalence and incidence of interstitial lung diseases in a multi- ethnic county of Greater Paris. Eur Respir J 2017;50(2).
5. Arkema EV, Grunewald J, Kullberg S, Eklund A, Askling J. Sarcoidosis incidence and prevalence: a nationwide register- based assessment in Sweden. Eur Respir J 2016;48:1690‑9.
6. Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc 2016;13:1244‑52.
7. Yoon HY, Kim HM, Kim YJ, Song JW. Prevalence and incidence of sarcoidosis in Korea: a nationwide population-based study. Respir Res 2018;19:158.
8. Cozier YC, Govender P, Berman JS. Obesity and sarcoidosis: consequence or contributor? Curr Opin Pulm Med 2018;24:487‑94.
9. Rossides M, Grunewald J, Eklund A, et al. Familial aggregation and heritability of sarcoidosis: a Swedish nested case- control study. Eur Respir J 2018;52(2).
10. Broos CE, van Nimwegen M, Hoogsteden HC, Hendriks RW, Kool M, van den Blink B. Granuloma formation in pulmonary sarcoidosis. Front Immunol 2013;4:437.
11. Ungprasert P, Crowson CS, Matteson EL. Seasonal variation in incidence of sarcoidosis: a population-based study 1976–2013. Thorax 2016;71:1164‑6.
12. Hena KM, Yip J, Jaber N, et al. Clinical course of sarcoidosis in world trade center-exposed firefighters. Chest 2018;153:114‑23.
13. Esteves T, Aparicio G, Garcia-Patos V. Is there any association between Sarcoidosis and infectious agents?: a systematic review and meta-analysis. BMC Pulm Med 2016;16:165.
14. Beghè D, Dall’Asta L, Garavelli C, et al. Sarcoidosis in an Italian province. Prevalence and environmental risk factors. PloS One 2017;12:e0176859.
15. Chen ES. Innate immunity in sarcoidosis pathobiology. Curr Opin Pulm Med 2016;22:469‑75.
16. Judson MA, Thompson BW, Rabin DL, et al. The diagnostic pathway to sarcoidosis. Chest 2003;123:406‑12.
17. Mañá J, Rubio-Rivas M, Villalba N, et al. Multidisciplinary approach and long-term follow-up in a series of 640 consecutive patients with sarcoidosis: Cohort study of a 40-year clinical experience at a tertiary referral center in Barcelona, Spain. Medicine (Baltimore) 2017;96:e7595.
18. Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med 2001;164(10 Pt 1):1885‑9.
19. Grunewald J, Eklund A. Sex-specific manifestations of Löfgren’s syndrome. Am J Respir Crit Care Med 2007;175:40‑4.
20. Drent M, Lower EE, De Vries J. Sarcoidosis-associated fatigue. Eur Respir J 2012;40:255‑63.
21. Bickett AN, Lower EE, Baughman RP. Sarcoidosis diagnostic score: a systematic evaluation to enhance the diagnosis of sarcoidosis. Chest 2018;154:1052-60.
22. Boussouar S, Medjhoul A, Bernaudin JF, et al. Diagnostic efficacy of ultrasound-guided core-needle biopsy of peripheral lymph nodes in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2015;32:188‑93.
23. Sehgal IS, Dhooria S, Aggarwal AN, Agarwal R. Impact of rapid on-site cytological evaluation (rose) on the diagnostic yield of transbronchial needle aspiration during mediastinal lymph node sampling: systematic review and meta-analysis. Chest 2017;153:929-38.
24. Pajares V, Puzo C, Castillo D, et al. Diagnostic yield of transbronchial cryobiopsy in interstitial lung disease: a randomized trial. Respirol Carlton Vic 2014;19:900‑6.
25. Hendriks C, Drent M, Elfferich M, De Vries J. The Fatigue Assessment Scale: quality and availability in sarcoidosis and other diseases. Curr Opin Pulm Med 2018;24:495‑503.
26. Schupp JC, Freitag-Wolf S, Bargagli E, et al. Phenotypes of organ involvement in sarcoidosis. Eur Respir J 2018;51(1).
27. Mahévas M, Chiche L, Uzunhan Y, et al. Association of sarcoidosis and immune thrombocytopenia: presentation and outcome in a series of 20 patients. Medicine (Baltimore) 2011;90:269‑78.
28. Piekarski E, Benali K, Rouzet F. Nuclear imaging in sarcoidosis. Semin Nucl Med 2018;48:246‑60.
29. Boucly A, Cottin V, Nunes H, et al. Management and long-term outcomes of sarcoidosis-associated pulmonary hypertension. Eur Respir J 2017;50(4).
30. Jamilloux Y, Maucort-Boulch D, Kerever S, et al. Sarcoidosis-related mortality in France: a multiple-cause-of-death analysis. Eur Respir J 2016;48:1700‑9.
31. Nardi A, Brillet PY, Letoumelin P, et al. Stage IV sarcoidosis: comparison of survival with the general population and causes of death. Eur Respir J 2011;38:1368‑73.
32. Nunes H, Uzunhan Y, Gille T, Lamberto C, Valeyre D, Brillet PY. Imaging of sarcoidosis of the airways and lung parenchyma and correlation with lung function. Eur Respir J 2012;40:750‑65.
33. Grenier P, Chevret S, Beigelman C, Brauner MW, Chastang C, Valeyre D. Chronic diffuse infiltrative lung disease: determination of the diagnostic value of clinical data, chest radiography, and CT and Bayesian analysis. Radiology 1994;191:383‑90.
34. Müller NL, Kullnig P, Miller RR. The CT findings of pulmonary sarcoidosis: analysis of 25 patients. Am J Roentgenol 1989;152:1179‑82.
35. Martin SG, Kronek LP, Valeyre D, et al. High-resolution computed tomography to differentiate chronic diffuse interstitial lung diseases with predominant ground-glass pattern using logical analysis of data. Eur Radiol 2010;20:1297‑310.
36. De Margerie-Mellon C, Dion G, Darlay J, et al. High-resolution computed tomography to differentiate chronic diffuse infiltrative lung diseases with chronic multifocal consolidation patterns using logical analysis of data. Sarcoidosis Vasc Diffuse Lung Dis 2016;33:355‑71.
37. Abehsera M, Valeyre D, Grenier P, Jaillet H, Battesti JP, Brauner MW. Sarcoidosis with pulmonary fibrosis: CT patterns and correlation with pulmonary function. Am J Roentgenol 2000;174:1751‑7.
38. Mihailovic-Vucinic V, Jovanovic D. Pulmonary sarcoidosis. Clin Chest Med 2008;29:459‑73.
39. Nunes H, Uzunhan Y, Freynet O, et al. Pulmonary hypertension complicating sarcoidosis. Presse Med 2012;41:e303‑16.
40. Baughman RP, Shlobin OA, Wells AU, et al. Clinical features of sarcoidosis associated pulmonary hypertension: Results of a multi-national registry. Respir Med 2018;139:72‑8.
41. Baughman RP, Culver DA, Cordova FC, et al. Bosentan for sarcoidosis-associated pulmonary hypertension: a double-blind placebo controlled randomized trial. Chest 2014;145:810‑7.
42. Denning DW, Pleuvry A, Cole DC. Global burden of chronic pulmonary aspergillosis complicating sarcoidosis. Eur Respir J 2013;41:621‑6.
43. Uzunhan Y, Nunes H, Jeny F, et al. Chronic pulmonary aspergillosis complicating sarcoidosis. Eur Respir J 2017;49(6).
44. Naccache JM, Lavolé A, Nunes H, et al. High-resolution computed tomographic imaging of airways in sarcoidosis patients with airflow obstruction. J Comput Assist Tomogr 2008;32:905‑12.
45. Birnie DH, Sauer WH, Judson MA. Consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Br Card Soc 2016;102:411‑4.
46. Chiu CZ, Nakatani S, Zhang G, et al. Prevention of left ventricular remodeling by long-term corticosteroid therapy in patients with cardiac sarcoidosis. Am J Cardiol 2005;95:143‑6.
47. Judson MA, Costabel U, Drent M, et al. The WASOG Sarcoidosis Organ Assessment Instrument: An update of a previous clinical tool. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:19‑27.
48. Terasaki F, Yoshinaga K. New guidelines for diagnosis of cardiac sarcoidosis in Japan. Ann Nucl Cardiol 2017;3:42‑5.
49. Birnie DH, Sauer WH, Bogun F, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm 2014;11:1305‑23.
50. Cohen Aubart F, Galanaud D, Haroche J, et al. Les atteintes neurologiques au cours de la sarcoïdose : diagnostic et traitement. Rev Med Interne 2017;38:393‑401.
51. Jachiet V, Lhote R, Rufat P, et al. Clinical, imaging, and histological presentations and outcomes of stroke related to sarcoidosis. J Neurol 2018;256:2333-41.
52. Stern BJ, Royal W, Gelfand JM, et al. Definition and consensus diagnostic criteria for neurosarcoidosis: from the Neurosarcoidosis Consortium Consensus Group. JAMA Neurol 2018;75:1546-53.
53. Ungprasert P, Wetter DA, Crowson CS, Matteson EL. Epidemiology of cutaneous sarcoidosis, 1976-2013: a population-based study from Olmsted County, Minnesota. J Eur Acad Dermatol Venereol 2016;30:1799‑804.
54. Noe MH, Rosenbach M. Cutaneous sarcoidosis. Curr Opin Pulm Med 2017;23:482‑6.
55. Kluger N. Sarcoidosis on tattoos: a review of the literature from 1939 to 2011. Sarcoidosis Vasc Diffuse Lung Dis 2013;30:86‑102.
56. Sève P, Kodjikian L, Jamilloux Y. Manifestations ophtalmologiques de la sarcoidose : que doit savoir l’interniste ? Rev Med Interne 2018;39:728‑37.
57. Herbort CP, Rao NA, Mochizuki M, members of Scientific Committee of First International Workshop on Ocular Sarcoidosis. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop On Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm 2009;17:160‑9.
58. Rothova A, Alberts C, Glasius E, Kijlstra A, Buitenhuis HJ, Breebaart AC. Risk factors for ocular sarcoidosis. Doc Ophthalmol Adv Ophthalmol 1989;72:287‑96.
59. Rochepeau C, Jamilloux Y, Kerever S, et al. Long-term visual and systemic prognoses of 83 cases of biopsy- proven sarcoid uveitis. Br J Ophthalmol 2017;101:856‑61.
60. Deutsch-Link S, Fortuna D, Weinberg EM. A comprehensive review of hepatic sarcoid. Semin Liver Dis 2018;38:284‑97.
61. Aubart FC, Ouayoun M, Brauner M, et al. Sinonasal involvement in sarcoidosis: a case-control study of 20 patients. Medicine (Baltimore) 2006;85:365‑71.
62. Duchemann B, Lavolé A, Naccache JM, et al. Laryngeal sarcoidosis: a case- control study. Sarcoidosis Vasc Diffuse Lung Dis 2014;31:227‑34.
63. Bergner R, Hoffmann M, Waldherr R, Uppenkamp M. Frequency of kidney disease in chronic sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2003;20:126‑32.
64. Cohen Aubart F, Abbara S, Maisonobe T, Cottin V, Papo T, Haroche J, et al. Symptomatic muscular sarcoidosis: Lessons from a nationwide multicenter study. Neurol Neuroimmunol Neuroinflammation 2018;5:e452.
65. Judson MA. The clinical features of sarcoidosis: a comprehensive review. Clin Rev Allergy Immunol 2015;49:63‑78.
66. Tavee JO, Karwa K, Ahmed Z, Thompson N, Parambil J, Culver DA. Sarcoidosis-associated small fiber neuropathy in a large cohort: Clinical aspects and response to IVIG and anti-TNF alpha treatment. Respir Med 2017;126:135‑8.
67. Heij L, Niesters M, Swartjes M, et al. Safety and efficacy of ARA 290 in sarcoidosis patients with symptoms of small fiber neuropathy: a randomized, double-blind pilot study. Mol Med Camb Mass 2012;18:1430‑6.
68. Wu CH, Chung PI, Wu CY, et al. Comorbid autoimmune diseases in patients with sarcoidosis: A nationwide case-control study in Taiwan. J Dermatol 2017;44:423‑30.
69. Bosse-Henck A, Koch R, Wirtz H, Hinz A. Fatigue and excessive daytime sleepiness in sarcoidosis: prevalence, predictors, and relationships between the two symptoms. Respir Int Rev Thorac Dis 2017;94:186‑97.
70. Reich JM. Mortality of intrathoracic sarcoidosis in referral vs population- based settings*: influence of stage, ethnicity, and corticosteroid therapy. CHEST J 2002;121:32.
71. Gribbin J, Hubbard RB, Le Jeune I, Smith CJP, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax 2006;61:980‑5.
72. Rossides M, Kullberg S, Askling J, Eklund A, Grunewald J, Arkema EV. Sarcoidosis mortality in Sweden: a population-based cohort study. Eur Respir J 2018;51(2).
73. Walsh SL, Wells AU, Sverzellati N, et al. An integrated clinicoradiological staging system for pulmonary sarcoidosis: a case-cohort study. Lancet Respir Med 2014;2:123‑30.
74. Kirkil G, Lower EE, Baughman RP. Predictors of mortality in pulmonary sarcoidosis. Chest 2018;153:105‑13.
75. Broos CE, Poell LHC, Looman CWN, et al. No evidence found for an association between prednisone dose and FVC change in newly-treated pulmonary sarcoidosis. Respir Med 2018;138S:S31‑7.
76. Broos CE, Wapenaar M, Looman CWN, et al. Daily home spirometry to detect early steroid treatment effects in newly treated pulmonary sarcoidosis. Eur Respir J 2018;51(1).
77. James WE, Baughman R. Treatment of sarcoidosis: grading the evidence. Expert Rev Clin Pharmacol 2018;11:677‑87.
78. Schutt AC, Bullington WM, Judson MA. Pharmacotherapy for pulmonary sarcoidosis: a Delphi consensus study. Respir Med 2010;104:717‑23.
79. Strookappe B, Swigris J, De Vries J, Elfferich M, Knevel T, Drent M. Benefits of physical training in sarcoidosis. Lung 2015;193:701‑8.
80. Taimeh Z, Hertz MI, Shumway S, Pritzker M. Lung transplantation for pulmonary sarcoidosis. Twenty-five years of experience in the USA. Thorax 2016;71:378‑9.
81. Zappala CJ, Desai SR, Copley SJ, et al. Accuracy of individual variables in the monitoring of long-term change in pulmonary sarcoidosis as judged by serial high-resolution CT scan data. Chest 2014;145:101‑7.
Dans cet article
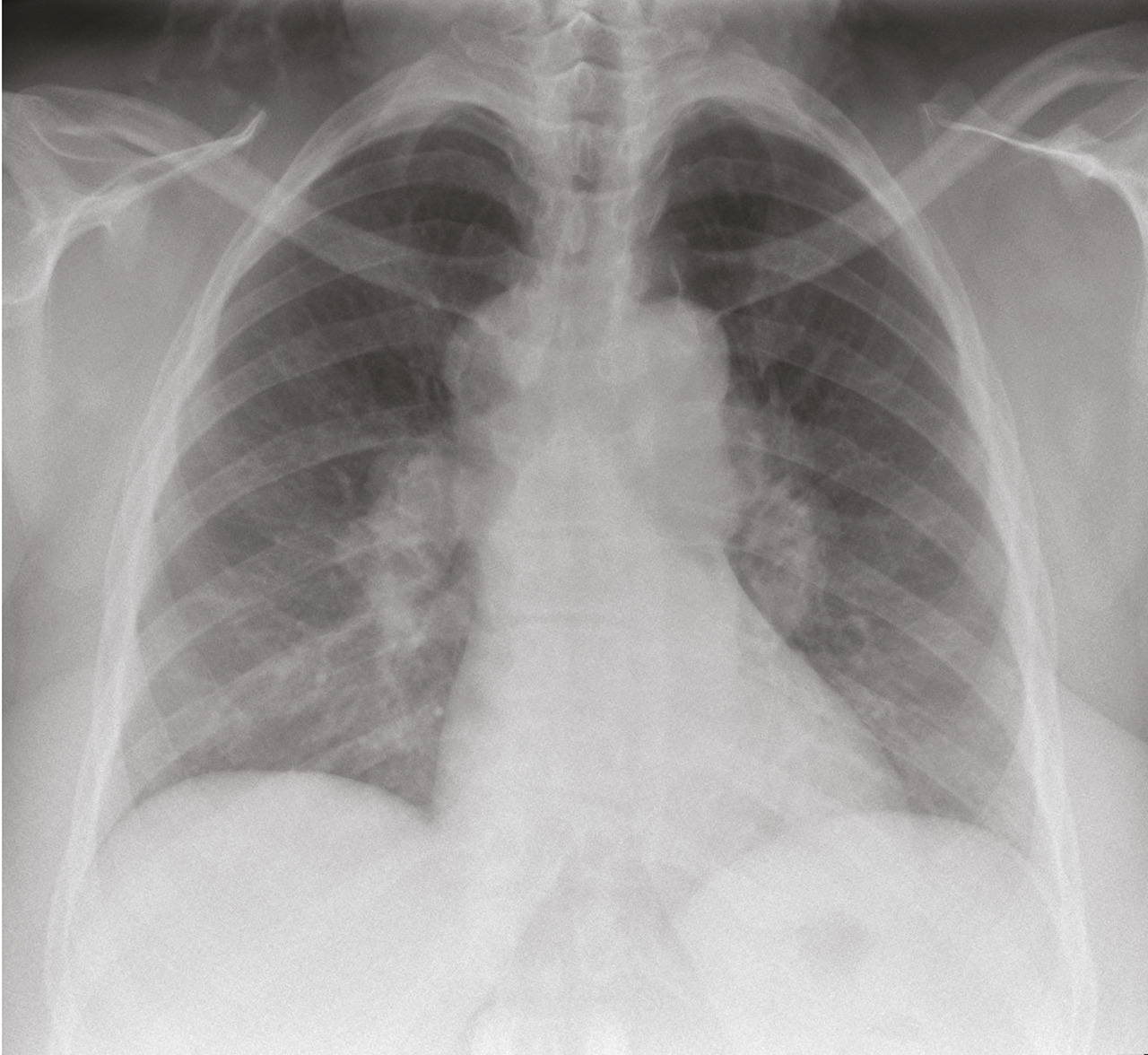
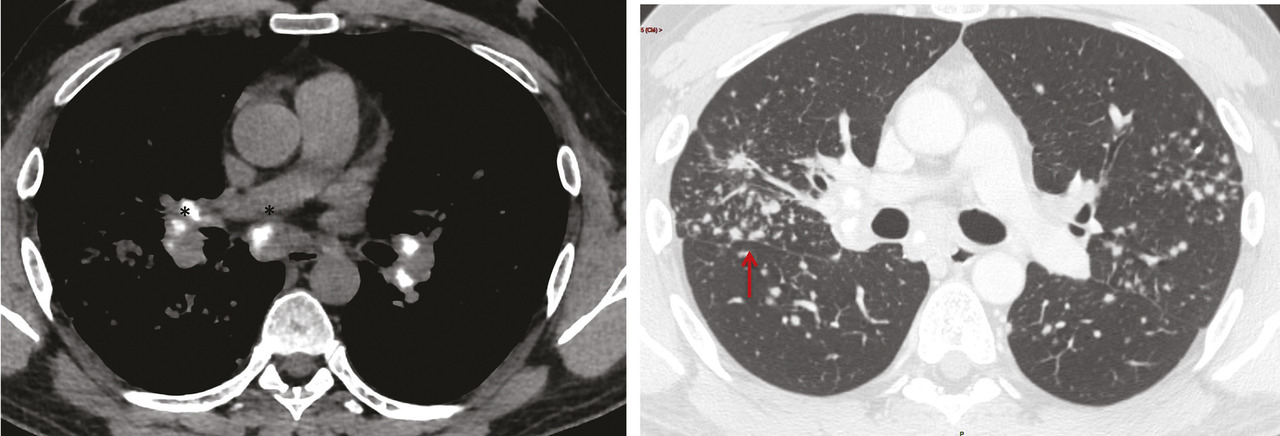
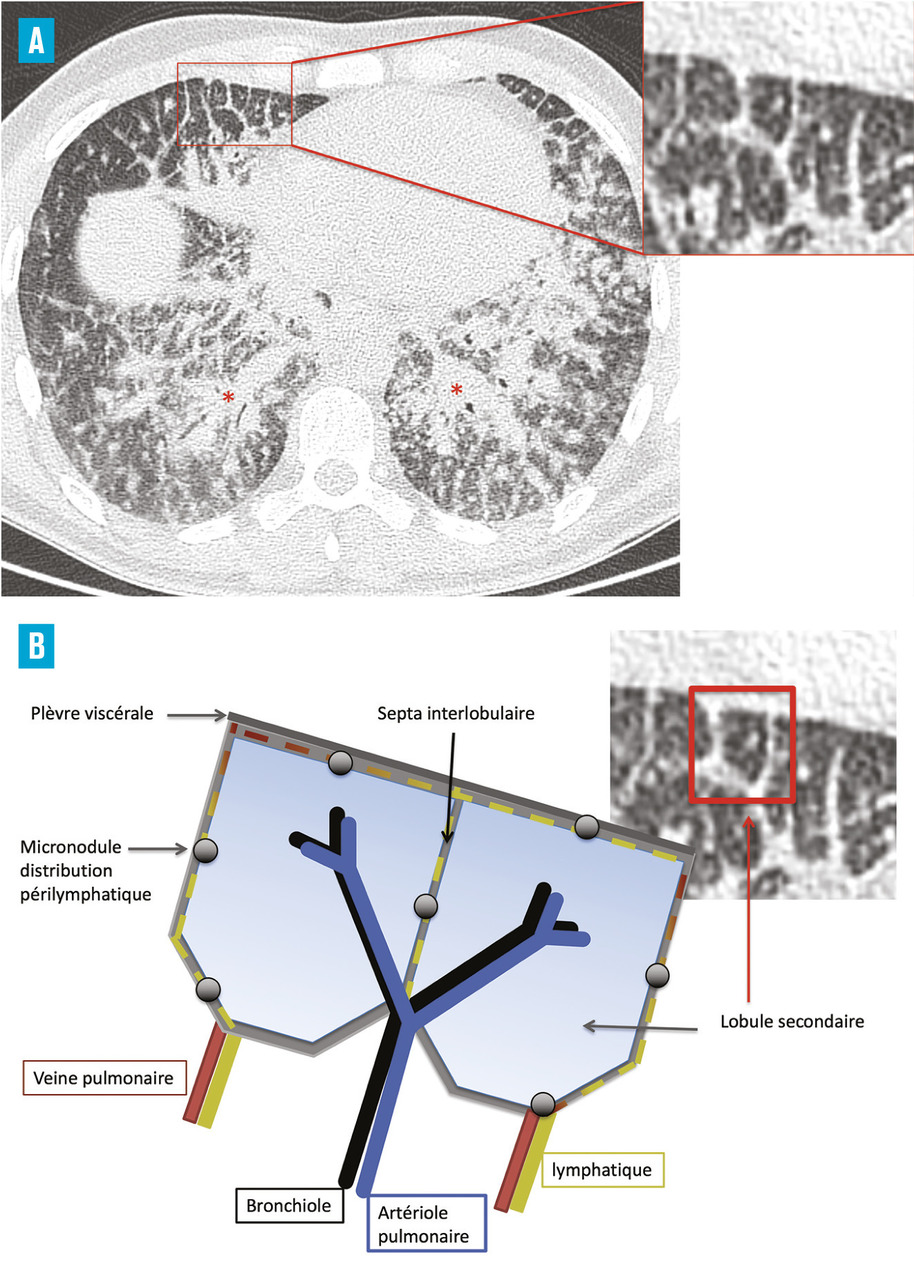
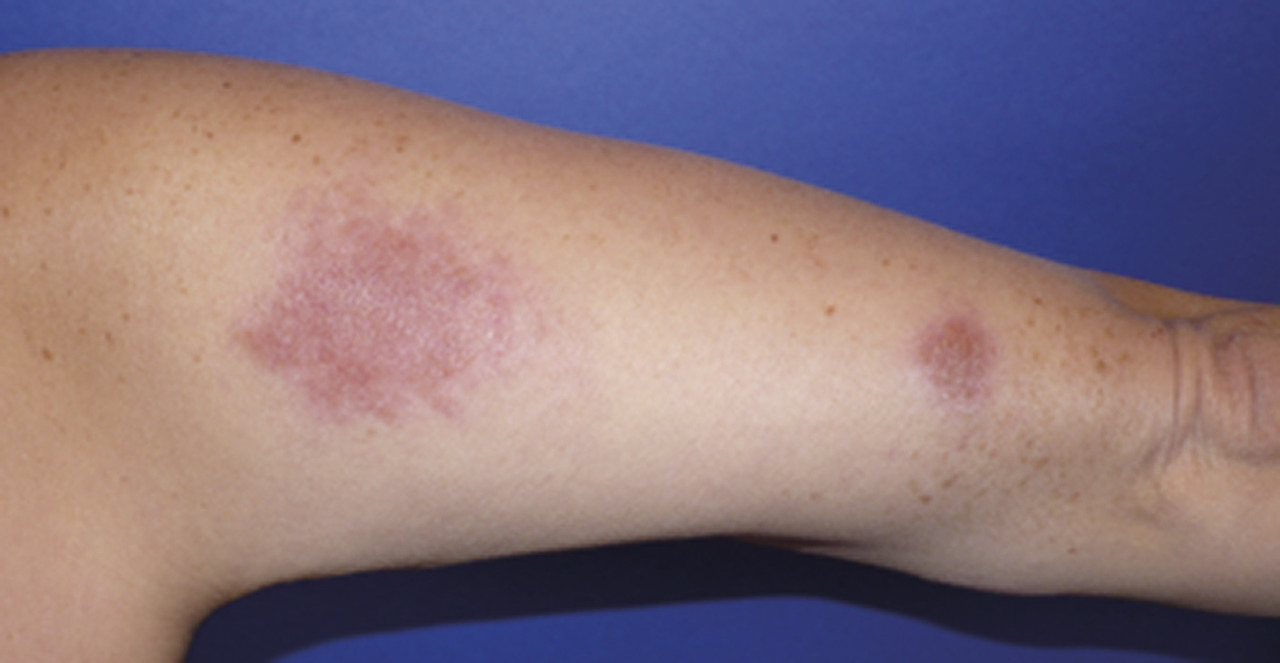
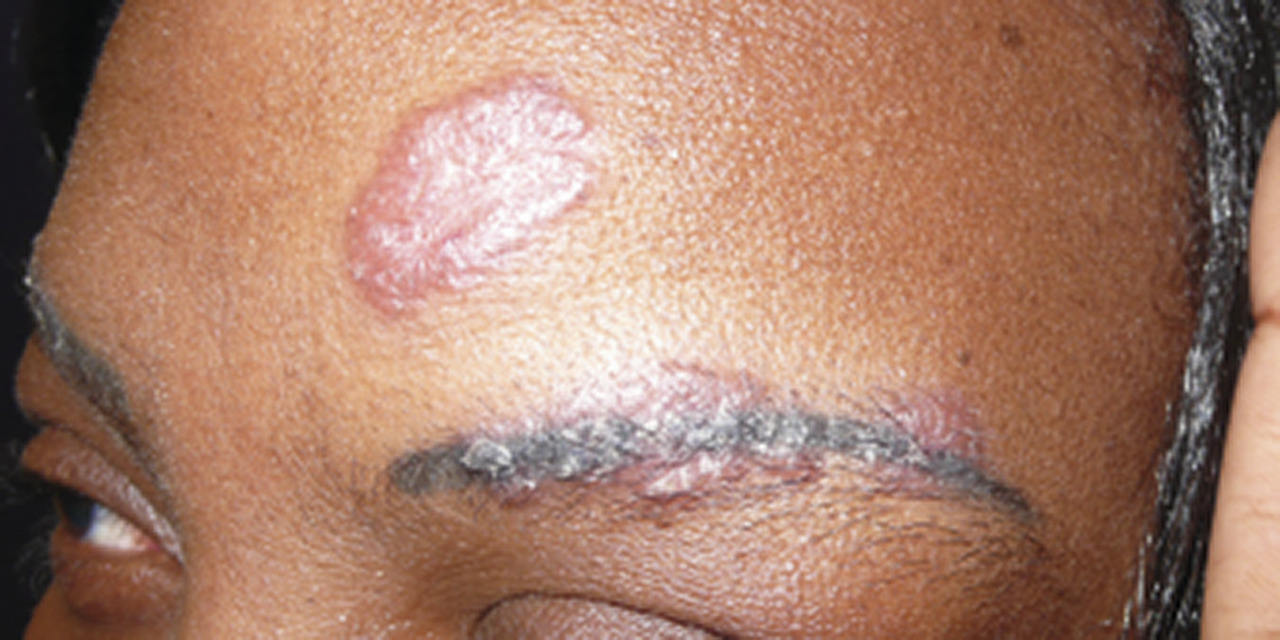


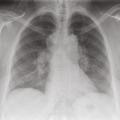
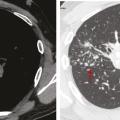
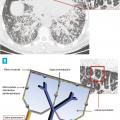
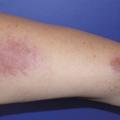
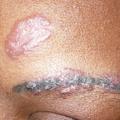
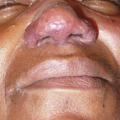
Résumé Sarcoïdose
La sarcoïdose est une maladie rare, de type granulomatose systémique de cause inconnue touchant presque constamment le poumon et le système lymphatique. Le diagnostic et la prise en charge thérapeutique peuvent être difficiles du fait de la diversité de ses présentations cliniques, du retentissement et de l’évolution de la maladie. Les objectifs du clinicien sont de ne pas manquer les formes avec dysfonction d’un organe pouvant mettre en jeu le pronostic vital (atteinte pulmonaire, cardiaque) ou fonctionnel (formes neurologique, ophtalmique, rénale), qualifiées de « dangereuses » mais aussi d’évaluer et prendre en charge les signes d’inconfort persistants, qui peuvent retentir sévèrement sur la qualité de vie des patients. Le traitement de la sarcoïdose n’est pas systématique mais, si nécessaire, les corticoïdes, aux effets indésirables notables, sont à donner en première intention. Les traitements de deuxième et de troisième ligne sont respectivement les immunosuppresseurs et les anti-tumor necrosis factor alpha.