La sclérodermie systémique est une connectivite touchant principalement les femmes (environ 80 % des cas) et débutant autour de 45 ans en moyenne. C’est une maladie rare, avec une prévalence en Europe entre 50 et 150 cas par million d’habitants. Le nombre total de patients en France est estimé entre 6 000 et 9 000. La physiopathologie, encore imparfaitement comprise, est complexe ; elle associe une vasculopathie prédominant sur la microcirculation, des dépôts excessifs de matrice extracellulaire et des mécanismes d’auto-immunité. Le pronostic est lié à la sévérité des atteintes d’organes : taux de survie à dix ans de 60 % pour les formes cutanées diffuses et de 80 % pour les formes cutanées limitées. Une prise en charge en centre expert dans la maladie est nécessaire.
Manifestations cliniques : hétérogènes et sévères
Les atteintes d’organes sont liées à deux mécanismes physiopathologiques : vasculopathie et fibrose par dépôts excessifs de matrice extracellulaire. Leur survenue est plus fréquente chez les patients atteints d’une forme diffuse de la maladie (tableau 1 ).
Atteintes par vasculopathie
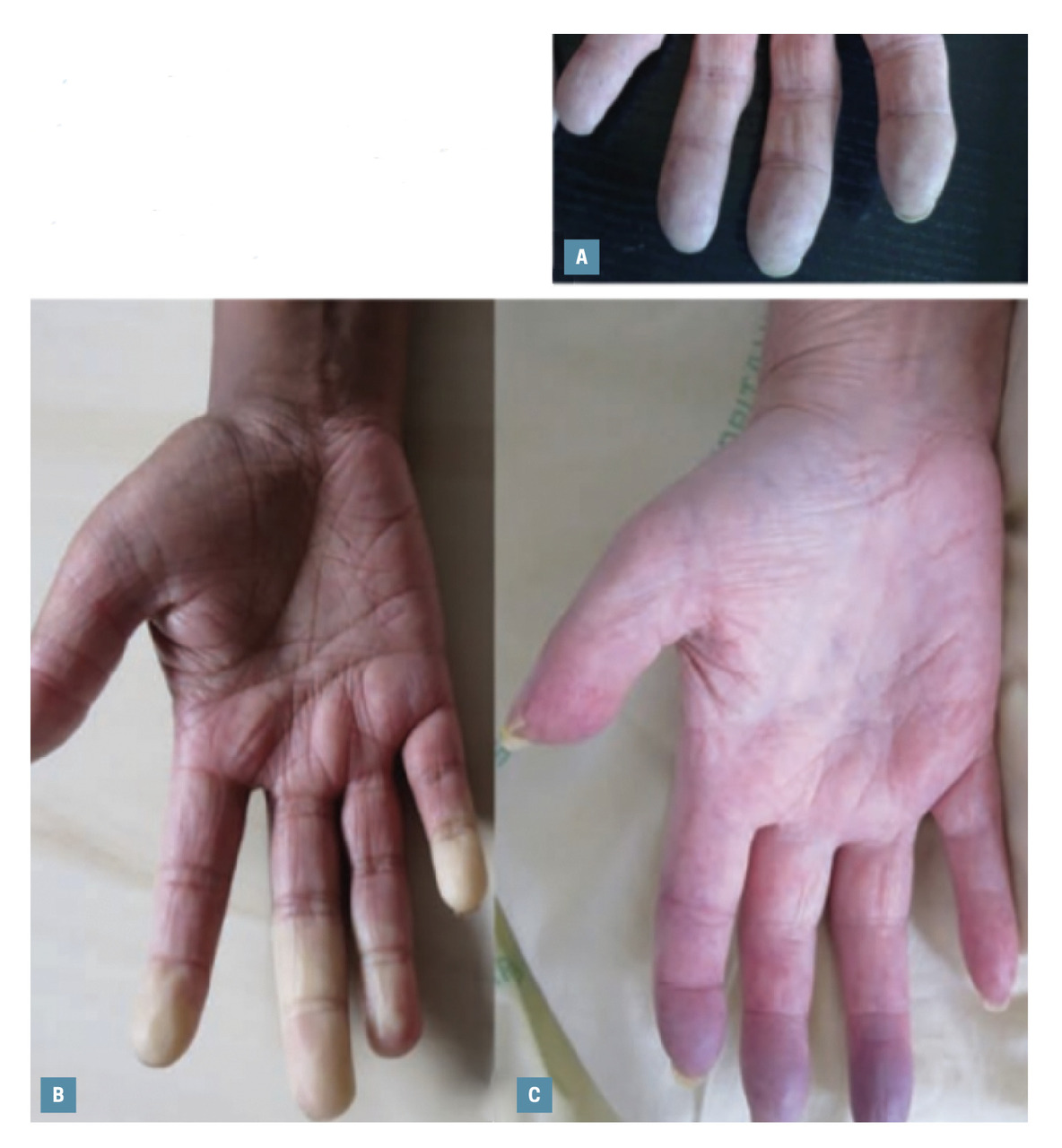
Phénomène de Raynaud
Le phénomène de Raynaud est quasiment constant. Il constitue la première manifestation de la maladie chez la majorité des patients, et apparaît souvent plusieurs années avant les autres signes.Il est essentiel d’en repérer précocement les caractéristiques cliniques sévères faisant suspecter une sclérodermie et le distinguant du phénomène de Raynaud idiopathique bénin : absence de rémission estivale, ulcérations digitales, phénomène affectant tous les doigts (pouces compris) et parfois les pieds, le nez, la langue et les oreilles. Deux réponses positives aux trois questions suivantes permettent d’éliminer les diagnostics différentiels que sont l’acrocyanose et la pâleur diffuse par vasoconstriction liée au froid : « Vos doigts sont-ils sensibles au froid ? », « Changent-ils de couleur avec le froid ? », « Deviennent-ils blancs puis bleus ? » (
Atteinte rénale
La crise rénale sclérodermique est une complication grave, caractérisée par une hypertension artérielle maligne avec insuffisance rénale aiguë oligurique. Elle concerne moins de 5 % des patients et apparaît précocement dans l’histoire de la maladie. La mortalité induite est importante : entre 18 et 36 % la première année.Hypertension artérielle pulmonaire
L’hypertension artérielle pulmonaire (HTAP) est également une complication grave, qui concerne 5 à 10 % des patients. Asymptomatique au stade précoce (absence de dyspnée, de douleur thoracique, de palpitations), elle doit faire l’objet d’un dépistage systématique car tout retard diagnostique grève le pronostic vital.Atteintes par fibrose
La peau : forme limitée ou diffuse
La fibrose cutanée donne son nom à la pathologie (aspect scléreux de la peau infiltrée par l’excès de dépôt de collagène).Selon l’étendue de l’atteinte cutanée, on distingue les formes dites « cutanées limitées », où la sclérose atteint les extrémités des membres sans remonter au-dessus des coudes et des genoux, et les formes « cutanées diffuses », où l’ensemble des téguments est touché. Le visage peut être concerné dans les deux formes. Les atteintes diffuses peuvent se compliquer de limitations d’amplitudes articulaires dans les cas les plus sévères. Outre la gravité cutanée, cette distinction entre formes limitées et diffuses est également importante sur le plan pronostique : les patients atteints de formes diffuses développent plus fréquemment des atteintes d’organes, et leur pronostic est donc plus altéré. Enfin, la peau peut être le siège de troubles de pigmentation parfois sévères et de télangiectasies, dont le préjudice esthétique peut être majeur.
Pneumopathie infiltrante diffuse : de mauvais pronostic
La pneumopathie infiltrante diffuse, peu symptomatique au stade précoce, est ensuite marquée par l’apparition d’une dyspnée d’effort, parfois d’une toux sèche, et de crépitants de type Velcro à l’auscultation pulmonaire. Elle peut évoluer vers l’insuffisance respiratoire chronique, et constitue l’une des principales causes de mortalité de la sclérodermie.Manifestations digestives fréquentes
La principale manifestation digestive de la sclérodermie est le reflux gastro-œsophagien, présent chez 80 à 90 % des patients, souvent invalidant, et pouvant se compliquer d’œsophagite peptique. Des troubles de la motilité intestinale sont également observés. Tous les segments du tube digestif peuvent être touchés. Une incontinence anale doit être recherchée à l’interrogatoire, car elle est fréquente et n’est pas toujours spontanément évoquée par les patients.Autres atteintes
Des atteintes musculo-squelettiques sont possibles : myalgies, arthralgies, frictions tendineuses, limitations de la mobilité articulaire.
Le cœur, enfin, peut être touché, avec l’apparition de troubles du rythme et de la conduction, une péricardite, voire une insuffisance cardiaque.
Le cœur, enfin, peut être touché, avec l’apparition de troubles du rythme et de la conduction, une péricardite, voire une insuffisance cardiaque.
Quels bilans réaliser ?
Les manifestations hétérogènes et les atteintes d’organes parfois peu symptomatiques aux stades initiaux rendent nécessaire une évaluation s’appuyant sur la clinique, et des examens complémentaires dès le diagnostic, puis au cours de l’évolution.
Confirmer le diagnostic
L’évaluation initiale cherche à confirmer le diagnostic chez les patients ayant une forme débutante. Des critères ont été établis à cette fin par l’American College of Rheumatology (ACR) et l’European League against Rheumatism (EULAR), en 2013 (tableau 2 ).
Ainsi, outre le recensement des manifestations cliniques, il convient de réaliser une capillaroscopie périunguéale (recherche de mégacapillaires associés à des plages avasculaires) et la recherche d’anticorps antinucléaires.
L’échographie cardiaque transthoracique avec mesure de la vitesse d’insuffisance tricuspide permet de rechercher uneHTAP. La tomodensitométrie thoracique en coupes fines à haute résolution est nécessaire pour objectiver une éventuelle pneumopathie infiltrante diffuse.
Associé au bilan biologique standard, le dosage des auto-anticorps est indispensable (tableau 2 ).
Les épreuves fonctionnelles respiratoires mesurent les débits et volumes pulmonaires ainsi que le coefficient de diffusion du monoxyde de carbone (DLCO).
Enfin, en cas de symptômes digestifs à type de reflux gastro-œsophagien sévère ou de dysphagie, une fibroscopie œsogastroduodénale est pratiquée.
Ainsi, outre le recensement des manifestations cliniques, il convient de réaliser une capillaroscopie périunguéale (recherche de mégacapillaires associés à des plages avasculaires) et la recherche d’anticorps antinucléaires.
L’échographie cardiaque transthoracique avec mesure de la vitesse d’insuffisance tricuspide permet de rechercher uneHTAP. La tomodensitométrie thoracique en coupes fines à haute résolution est nécessaire pour objectiver une éventuelle pneumopathie infiltrante diffuse.
Associé au bilan biologique standard, le dosage des auto-anticorps est indispensable (
Les épreuves fonctionnelles respiratoires mesurent les débits et volumes pulmonaires ainsi que le coefficient de diffusion du monoxyde de carbone (DLCO).
Enfin, en cas de symptômes digestifs à type de reflux gastro-œsophagien sévère ou de dysphagie, une fibroscopie œsogastroduodénale est pratiquée.
Surveiller l’évolution
La surveillance des patients comprend une évaluation clinique et biologique annuelle. Les épreuves fonctionnelles respiratoires et l’échographie cardiaque transthoracique sont répétées à la même fréquence.
Les autres examens dépendent de la symptomatologie (tableau 3 ).
Les autres examens dépendent de la symptomatologie (
Prise en charge thérapeutique
La complexité physiopathologique et l’hétérogénéité des manifestions rendent la prise en charge complexe. Elle se décline en deux axes : traitement des complications d’organes et traitement de fond (tableau 4 ). Il n’existe pas de protocole standard applicable à tous les patients.
Traitement des complications viscérales
Le phénomène de Raynaud implique la mise en place de mesures préventives (éviction du froid et de l’humidité, arrêt du tabac, arrêt des bêtabloquants…). L’instauration d’un traitement par inhibiteur calcique est le plus souvent nécessaire. En cas d’ulcérations digitales, les analogues de prostacycline sont indiqués, et un inhibiteur des récepteurs à l’endothéline est utilisé en prévention des récidives.
Les inhibiteurs de l’enzyme de conversion, prescrits au décours de la crise rénale sclérodermique, permettent d’améliorer le pronostic. On envisage une épuration extrarénale dans les formes les plus graves, voire une transplantation rénale en cas d’insuffisance rénale terminale.
La prise en charge de l’HTAP dépend du stade de la dyspnée : antagoniste des récepteurs de l’endothéline et/ou inhibiteur de la phosphodiestérase 5 pour les stades 2 et 3, agonistes de la prostacycline en cas de stade 4. La transplantation cardiopulmonaire est proposée en ultime recours, en cas d’échec du traitement médicamenteux.
Le nintédanib, un antifibrosant utilisé dans la fibrose pulmonaire idiopathique, a récemment obtenu l’autorisation de mise sur le marché pour la pneumopathie infiltrante diffuse. Sa place dans la stratégie thérapeutique, jusqu’ici dominée par les immunosuppresseurs, reste à définir. En cas d’insuffisance respiratoire terminale, c’est la transplantation bipulmonaire qui est à envisager.
La prescription d’inhibiteurs de la pompe à protons à forte dose est recommandée en cas d’atteinte œsophagienne. En cas de gastroparésie, l’érythromycine prescrite à dose faible a un effet prokinétique.
Les corticoïdes à faible dose peuvent apporter une amélioration des signes articulaires, musculaires, cardiaques et pulmonaires ; en revanche, les fortes doses (> 15 mg/j) sont à éviter (risque accru de crise rénale sclérodermique).
Les atteintes cutanées diffuses relèvent des traitements de fond.
Les inhibiteurs de l’enzyme de conversion, prescrits au décours de la crise rénale sclérodermique, permettent d’améliorer le pronostic. On envisage une épuration extrarénale dans les formes les plus graves, voire une transplantation rénale en cas d’insuffisance rénale terminale.
La prise en charge de l’HTAP dépend du stade de la dyspnée : antagoniste des récepteurs de l’endothéline et/ou inhibiteur de la phosphodiestérase 5 pour les stades 2 et 3, agonistes de la prostacycline en cas de stade 4. La transplantation cardiopulmonaire est proposée en ultime recours, en cas d’échec du traitement médicamenteux.
Le nintédanib, un antifibrosant utilisé dans la fibrose pulmonaire idiopathique, a récemment obtenu l’autorisation de mise sur le marché pour la pneumopathie infiltrante diffuse. Sa place dans la stratégie thérapeutique, jusqu’ici dominée par les immunosuppresseurs, reste à définir. En cas d’insuffisance respiratoire terminale, c’est la transplantation bipulmonaire qui est à envisager.
La prescription d’inhibiteurs de la pompe à protons à forte dose est recommandée en cas d’atteinte œsophagienne. En cas de gastroparésie, l’érythromycine prescrite à dose faible a un effet prokinétique.
Les corticoïdes à faible dose peuvent apporter une amélioration des signes articulaires, musculaires, cardiaques et pulmonaires ; en revanche, les fortes doses (> 15 mg/j) sont à éviter (risque accru de crise rénale sclérodermique).
Les atteintes cutanées diffuses relèvent des traitements de fond.
Traitements de fond
Les traitements immunosuppresseurs et immunomodulateurs sont réservés aux sclérodermies systémiques diffuses de diagnostic antérieur à cinq ans ou évolutives : mycophénolate mofétil, cyclophosphamide, rituximab, anti-interleukine 6, etc.
Dans les formes très sévères et rapidement évolutives, après discussion en réunion de concertation pluridisciplinaire spécialisée, une intensification thérapeutique suivie d’une autogreffe de cellules souches hématopoïétiques peut être proposée.
Enfin, la prise en charge fonctionnelle est importante pour lutter contre le handicap induit par la maladie, notamment du fait de l’atteinte des mains.
Dans les formes très sévères et rapidement évolutives, après discussion en réunion de concertation pluridisciplinaire spécialisée, une intensification thérapeutique suivie d’une autogreffe de cellules souches hématopoïétiques peut être proposée.
Enfin, la prise en charge fonctionnelle est importante pour lutter contre le handicap induit par la maladie, notamment du fait de l’atteinte des mains.
Pour en savoir plus
1. HAS. Protocole national de diagnostic et de soins. Sclérodermie systémique. 2017 (révision janvier 2020). Disponible sur : https://bit.ly/3zzc8P4
2. Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2017;76(8):1327-39.
3. Denton CP, Khanna D. Systemic sclerosis. Lancet 2017;390(10103):1685-99.
4. Van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013;65(11):2737-47.
5. Legendre P, Mouthon L. Sclérodermie systémique. Rev Prat 2017;67(7):775-83.
2. Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2017;76(8):1327-39.
3. Denton CP, Khanna D. Systemic sclerosis. Lancet 2017;390(10103):1685-99.
4. Van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013;65(11):2737-47.
5. Legendre P, Mouthon L. Sclérodermie systémique. Rev Prat 2017;67(7):775-83.
Dans cet article

essentiel
La sclérodermie systémique est une connectivite dont le tableau clinique est hétérogène, marqué par un retentissement important sur la qualité de vie et dont les atteintes d’organes peuvent engager le pronostic vital.
Le phénomène de Raynaud est un signe quasiment constant d’apparition très précoce. Son repérage doit inciter à rechercher des caractéristiques et des signes associés évocateurs de maladie systémique.
La prise en charge est complexe et individualisée. La coordination entre plusieurs spécialistes est le plus souvent nécessaire.