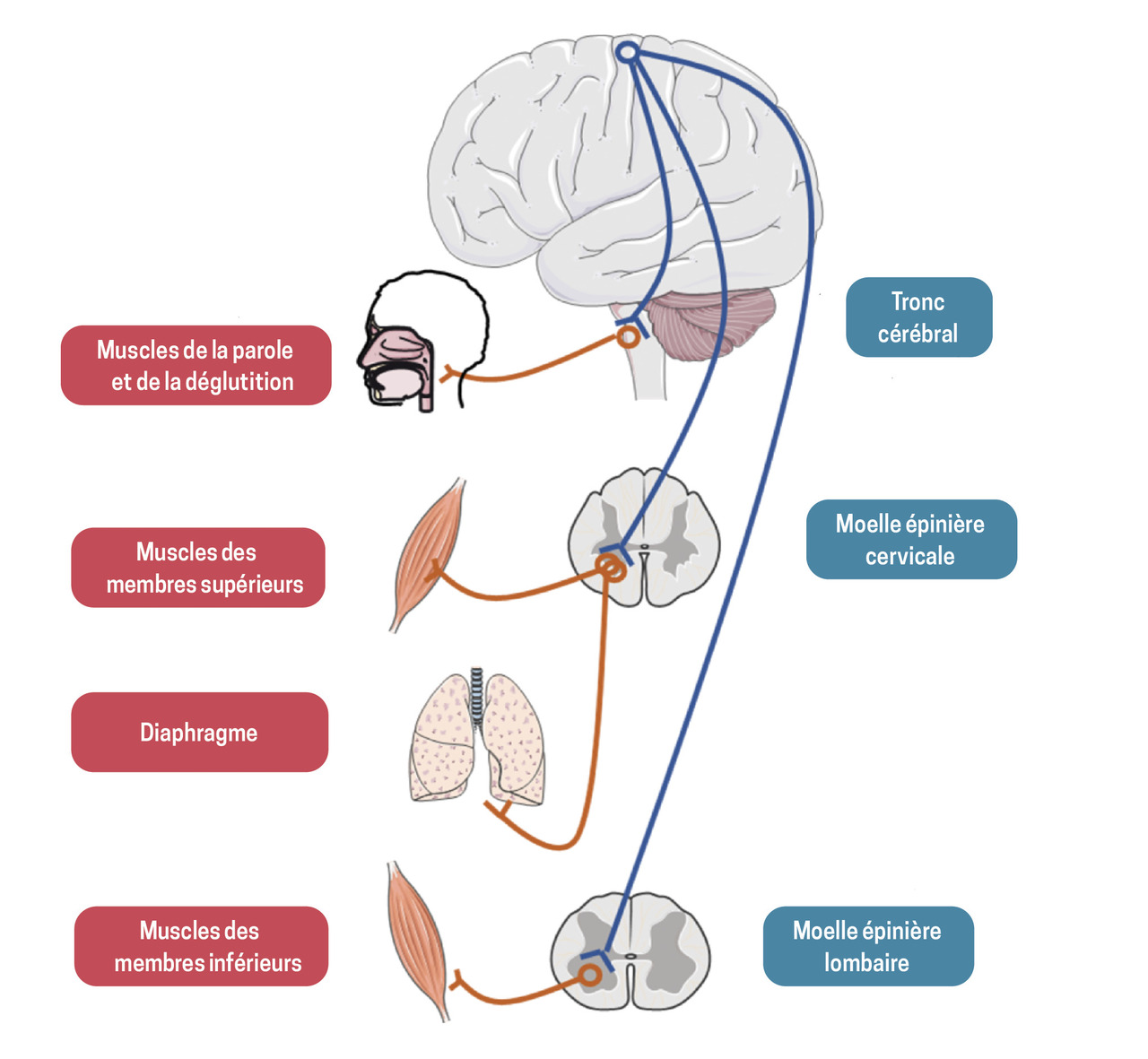
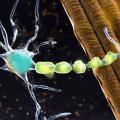
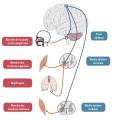
La sclérose latérale amyotrophique (SLA), également connue sous le nom de maladie de Charcot, est une pathologie neurodégénérative caractérisée par une atteinte des motoneurones du cortex cérébral (motoneurone central [MNC]), du bulbe et de la corne antérieure de la moelle épinière (motoneurone périphérique [MNP]) [fig. 1].
Bien qu’elle soit la maladie du motoneurone la plus fréquente chez l’adulte, elle reste relativement rare : son incidence en Europe est estimée entre 2 et 3 pour 100 000 habitants par an, et le risque global de développer une SLA au cours d’une vie est de 1/350 pour les hommes et 1/400 pour les femmes.1
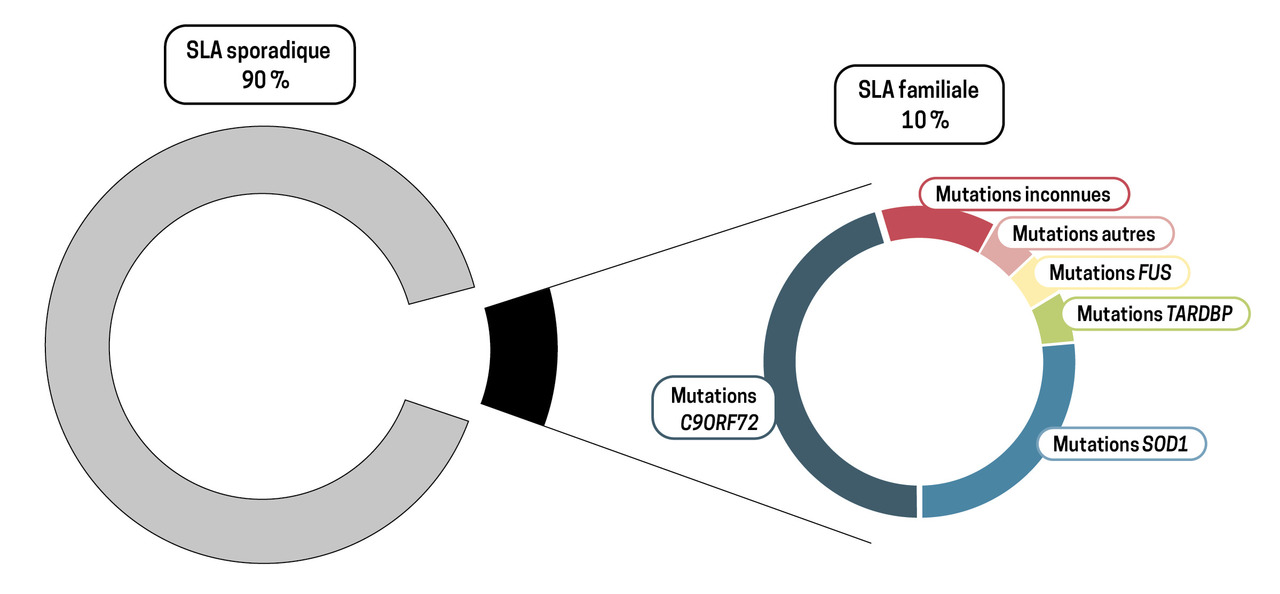
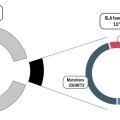
Dans la majorité des cas, la SLA survient de façon sporadique ; il existe néanmoins une histoire familiale de la maladie pour environ 10 % des patients, liée dans 70 % des cas à une mutation de l’un des quatre gènes principaux responsables de la SLA : C9ORF72, SOD1, TARDBP ou FUS (fig. 2).
Si les connaissances sur la pathogénie de la SLA progressent, les mécanismes physiopathologiques en cause, qui font intervenir de multiples voies (anomalie de l’homéostasie protéique, métabolisme des ARN et des mitochondries, du stress oxydatif, perturbations du transport axonal et de la jonction neuromusculaire, rôle de la neuro-inflammation…), restent incomplètement élucidés.
Poser le diagnostic de SLA
L’absence de marqueurs sanguins rend le diagnostic difficile, notamment pour les formes débutantes, dans lesquelles les signes sont incomplets.
Des formes cliniques très variées
La SLA a une grande hétérogénéité clinique, aussi bien dans sa manifestation que dans son évolution. La maladie survient généralement vers l’âge de 65 ans,2 mais peut débuter chez des patients de moins de 20 ans et jusqu’à plus de 90 ans.
Le diagnostic repose essentiellement sur l’examen clinique, qui montre des signes d’atteinte des motoneurones, touchant de façon progressive les quatre territoires bulbaire, cervical, thoracique et lombaire.
La dégénérescence du MNC se traduit par un syndrome pyramidal, avec une parésie motrice sans amyotrophie et un ralentissement des mouvements fins. L’examen clinique peut objectiver une hypertonie spastique, une exagération des réflexes ostéotendineux, un signe de Babinski.
L’atteinte du MNP est caractérisée par une fatigabilité musculaire, des crampes, et l’examen retrouve des fasciculations, un déficit musculaire, une amyotrophie, une hypotonie et une diminution des réflexes ostéotendineux.
Typiquement, les patients n’ont pas de troubles sensitifs, sphinctériens ni oculomoteurs.
Au sein de ce phénotype classique de SLA, environ deux tiers des cas sont dits de forme à début spinal : l’atteinte motoneuronale se manifeste par un déficit moteur progressif amyotrophiant asymétrique des membres. Les formes débutant par une atteinte respiratoire sont beaucoup plus rares (moins de 3 % des cas) et peuvent être révélées par des signes chroniques d’hypoventilation alvéolaire ou par un tableau de décompensation respiratoire aiguë, le plus souvent hypercapnique.
À l’inverse, un tiers des patients ont une forme à début bulbaire, caractérisée par une dysarthrie et des troubles de déglutition qui prédominent souvent initialement sur les liquides.
Enfin, il existe une atteinte cognitive ou comportementale associée à l’atteinte motrice pour environ la moitié des patients,2 dont 5 à 15 % avec un véritable tableau de démence frontotemporale.
Examens paracliniques : pas de marqueur spécifique
En l’absence de marqueur diagnostique spécifique, le diagnostic de SLA repose sur « la mise en évidence de signes cliniques et électromyographiques d’atteinte du MNC et du MNP, sur le caractère évolutif des signes et l’absence d’éléments en faveur d’une autre pathologie pouvant expliquer les signes observés », d’après la conférence de consensus de la Haute Autorité de santé.3
Électroneuromyogramme, le gold standard
Lorsque le diagnostic est suspecté, l’électroneuromyogramme est l’examen de référence. Il permet de confirmer l’atteinte du motoneurone périphérique et d’en préciser l’extension.
IRM cérébrale et médullaire pour éliminer les diagnostics différentiels
Il est également recommandé de réaliser des IRM cérébrale et médullaire, dont l’objectif principal est d’éliminer un diagnostic différentiel (lésion du tronc cérébral, myélopathie cervicarthrosique).
Un hypersignal localisé sur le trajet du faisceau corticospinal en séquences T2 peut être mis en évidence sur l’IRM cérébrale.
Autres examens au cas par cas
En fonction du tableau clinique, des comorbidités associées et de l’évolution des symptômes, un complément d’exploration peut se justifier après avis spécialisé.
Évolution péjorative nécessitant un suivi rapproché
Le pronostic de la SLA est sombre, puisque le décès survient en moyenne dans les deux à cinq ans après l’apparition des symptômes. Il existe cependant des formes lentes de la maladie, avec des survies prolongées supérieures à dix ans.4
La dégénérescence des motoneurones entraîne une perte progressive des fonctions motrices, avec un retentissement fonctionnel variable selon les territoires atteints. L’atteinte de la sphère bulbaire est évaluée par le bilan de la parole, de la voix, des praxies bucco-faciales et de la déglutition, au mieux réalisé par un orthophoniste.
Dénutrition fréquente et multifactorielle
Le statut nutritionnel – évalué par la pente de perte de poids et le calcul de l’indice de masse corporelle – est un élément indispensable du suivi. En effet, une dénutrition est retrouvée chez jusqu’à 50 % des patients et est un facteur pronostique négatif indépendant pour la survie.5 Elle peut être multifactorielle : troubles de la déglutition, anorexie liée aux conséquences psychologiques de la maladie, hypermétabolisme avec augmentation de la dépense énergétique de repos.
Rechercher régulièrement l’apparition d’une atteinte respiratoire
L’évolution vers une atteinte respiratoire par insuffisance diaphragmatique est quasiment constante chez les patients atteints de SLA et fait toute la sévérité du pronostic. La recherche régulière d’une atteinte respiratoire est donc essentielle.
Les principaux symptômes évocateurs d’insuffisance diaphragmatique sont la dyspnée, l’orthopnée, les troubles du sommeil (réveils nocturnes, hypersomnolence diurne, céphalées matinales).
L’examen clinique recherche des signes de recrutement des muscles respiratoires accessoires et une respiration abdominale paradoxale en décubitus dorsal.
Les principaux examens permettant un suivi de la fonction respiratoire sont les gaz du sang artériels, les explorations fonctionnelles respiratoires et l’oxymétrie nocturne.
Quelques facteurs pronostiques identifiés
Outre les facteurs déjà évoqués, les principaux éléments pronostiques péjoratifs sont l’âge élevé au début des symptômes, une forme de début bulbaire ou respiratoire, un délai court entre le début des symptômes et le diagnostic (traduisant une évolution rapide) et une pente rapide de dégradation de la fonction musculaire.
Quelle prise en charge ?
Multidisciplinarité et coordination
L’annonce diagnostique est habituellement faite par le neurologue lors d’une consultation dédiée. Ce premier temps est un élément fondateur de la relation entre le patient et l’équipe le prenant en charge.
Les objectifs de la prise en charge sont multiples : suivi régulier, mise en place de traitements étiologiques et symptomatiques, prise en charge du handicap moteur et des fonctions vitales, soutien psychologique.
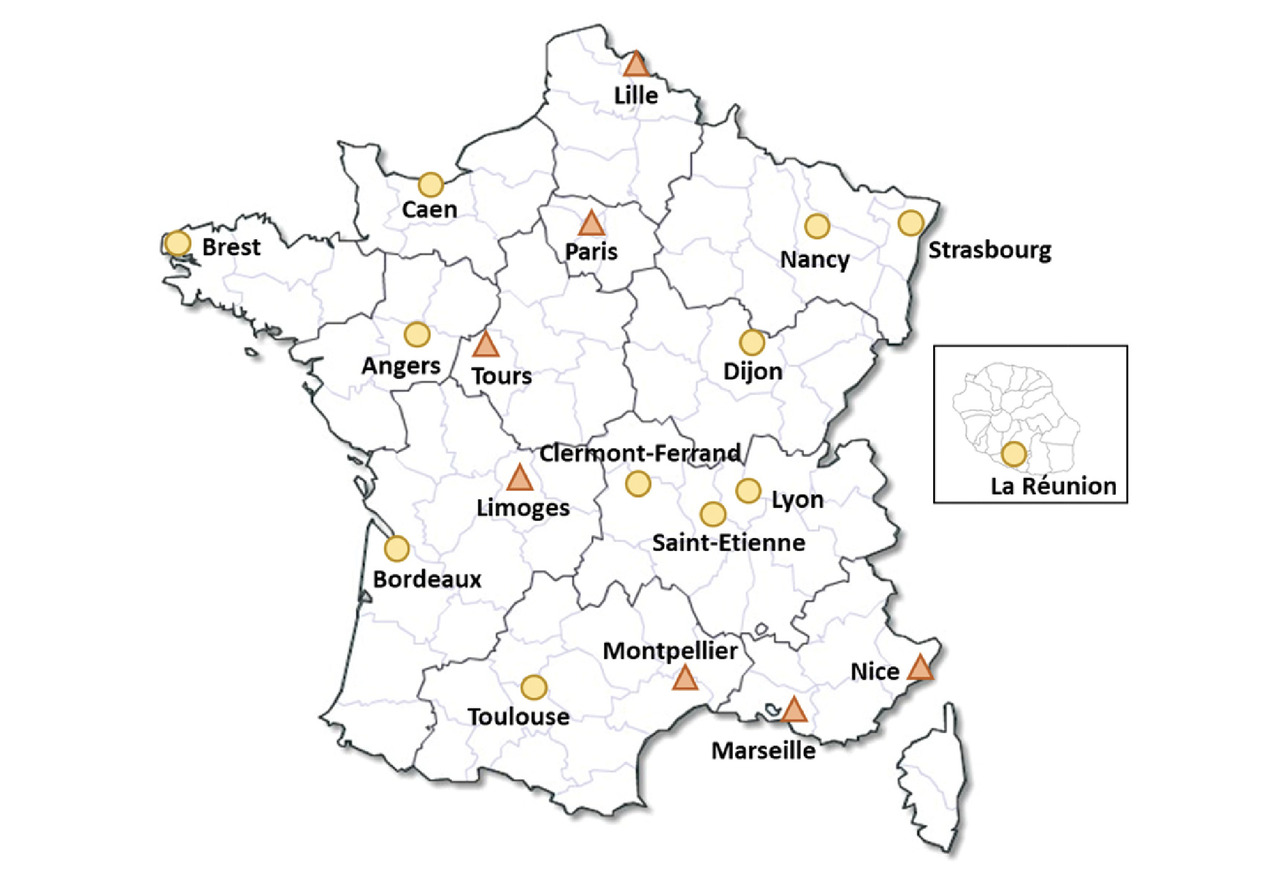
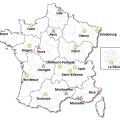
Cette prise en charge nécessite une multidisciplinarité et fait intervenir différentes professions médicales, paramédicales et du domaine du médico-social. Elle doit être, au mieux, coordonnée dans l’un des dix-neuf centres SLA français (fig. 3), en lien avec le médecin traitant.
Traitements médicamenteux
Il n’existe actuellement aucun traitement curatif de la SLA.
Une seule AMM en France
Le riluzole, traitement neuroprotecteur ciblant l’excitotoxicité des neurones, est le seul traitement ayant montré un effet modeste sur la survie des patients6 et le seul ayant obtenu une autorisation de mise sur le marché (AMM) en France. Il est instauré par un médecin expérimenté dans la prise en charge des maladies du motoneurone. Ses effets indésirables principalement rapportés sont une asthénie, des troubles digestifs, une cytolyse hépatique – nécessitant une surveillance régulière des transaminases – et une neutropénie – nécessitant la surveillance de l’hémogramme.
Édavarone
En 2018, l’édaravone par voie intraveineuse, ciblant le stress oxydatif, a obtenu une autorisation d’accès compassionnel (AAC) à la suite d’un essai de phase III réalisé dans une sous-population de patients ayant une forme « précoce » de SLA.7 Cependant, la nécessité de perfusions intraveineuses fréquentes (10 jours tous les 28 jours lors des cycles d’entretien) et les interrogations persistantes sur son efficacité – une étude récente n’ayant mis en évidence aucun effet sur la progression de la maladie8 – ont limité sa prescription en France. Une large étude multicentrique de phase III est en cours en Europe afin d’évaluer une formulation orale de l’édaravone.
Perspectives thérapeutiques
L’association phénylbutyrate de sodium-taurursodiol (AMX0035), ciblant le stress du réticulum endoplasmique et la dysfonction mitochondriale, a été approuvée en 2022 au Canada et aux États-Unis, suivant les résultats d’une étude de phase II ayant montré un ralentissement du déclin fonctionnel de la maladie.9 Une large étude internationale de phase III est en cours pour confirmer son efficacité, et l’Agence européenne du médicament devrait se prononcer courant 2023 sur son autorisation.
Une autre approche thérapeutique en plein essor est celle des thérapies ciblées sur des gènes responsables de la pathologie. Le tofersen (oligonucléotide antisens anti-SOD1), injecté mensuellement par voie intrathécale, a ainsi montré des résultats encourageants, suggérant une réduction du déclin des fonctions motrice et respiratoire10 et a obtenu en 2022 en France une AAC pour les patients atteints de SLA génétique liée à une mutation du gène SOD1.
Prise en charge de l’insuffisance respiratoire
L’avènement de la ventilation non invasive (VNI), méthode de référence de suppléance respiratoire dans la SLA, a permis d’améliorer significativement la survie et la qualité de vie des patients. Sa mise en place, sur des critères cliniques et/ou paracliniques d’insuffisance diaphragmatique, est habituellement réalisée par une équipe spécialisée de pneumologie. Elle est, en règle générale, débutée la nuit, mais la progression de la pathologie conduit à augmenter la dépendance à la VNI.
Lorsque la VNI ne suffit plus, la question d’une ventilation mécanique permanente par le biais d’une trachéotomie peut être posée, mais elle modifie fondamentalement le mode et la qualité de vie (soins itératifs d’aspirations trachéales ou de changement de canules qui peuvent être inconfortables, voire douloureux, problématiques de la perte de communication orale et de la déglutition, lourdeur des soins) et une très grande majorité de patients choisit de s’orienter plutôt vers des soins de confort lorsque la VNI devient insuffisante.
Dans tous les cas, le projet de recourir ou non à la ventilation par trachéotomie doit être anticipé et nécessite une multidisciplinarité et une collégialité dans l’échange et la réflexion sur les objectifs souhaités par le patient et son entourage.
Un autre élément essentiel est la prise en charge de l’encombrement des voies respiratoires lié à une toux inefficace, fréquemment rencontré chez les patients ayant une atteinte respiratoire et/ou bulbaire.
Les moyens utilisés sont multiples, souvent complémentaires : kinésithérapie respiratoire de désencombrement, mise en place d’un système de désencombrement au domicile (aspirateur à mucosités, in-exsufflateur mécanique), traitement médicamenteux pour lutter contre l’hypersialorrhée (hors AMM : scopolamine en patch transdermique, atropine en collyre sublingual…).
Prise en charge nutritionnelle
Chez les patients avec atteinte bulbaire, une prise en charge orthophonique est indispensable afin de lutter au mieux contre les troubles de la déglutition (apprentissage de postures pour faciliter le passage du bol alimentaire, adaptation des textures alimentaires).
Une prise en charge diététique avec éducation du patient à un régime alimentaire riche en protides et graisses, si besoin associé à la prescription de compléments nutritionnels oraux hypercaloriques, est aussi essentielle. Lorsque ces mesures ne permettent pas d’enrayer la perte de poids ou que l’alimentation devient impossible du fait de la progression de l’atteinte bulbaire, la mise en place d’une alimentation entérale par le biais d’une sonde de gastrostomie est la méthode de référence dans la SLA.
Une attention particulière doit être portée aux patients âgés ayant une dénutrition sévère ou une atteinte respiratoire, facteurs pronostiques pouvant influencer négativement les risques de complications post-gastrostomie. Là encore, la décision parfois complexe de réalisation d’une gastrostomie peut nécessiter une expertise et une multidisciplinarité des équipes soignantes prenant en charge le patient.
Prise en charge symptomatique
Le reste de la prise en charge est dominé par les traitements symptomatiques de la douleur, des crampes, de la spasticité, des troubles de l’humeur et des troubles anxieux induits par la pathologie, et des complications du décubitus.
Médecin généraliste : un rôle central
Si certaines de ces prises en charge nécessitent l’intervention de médecins spécialistes (neurologue, pneumologue, médecin spécialiste en médecine physique et réadaptation, gastroentérologue), le rôle du médecin traitant est central dans le suivi des patients, le dépistage des complications spécifiques (alerte sur le risque de dénutrition ou d’insuffisance respiratoire) ou liées à la perte d’autonomie (risque d’infection pulmonaire et risque thromboembolique, notamment).
Il est un interlocuteur médical privilégié pour le patient et son entourage et aide à faire le lien entre les différents intervenants médicaux et paramédicaux.
S’appuyer sur les intervenants paramédicaux et le psychologue
La rééducation en kinésithérapie (en cas de déficit moteur ou d’atteinte respiratoire) ou en orthophonie (en cas d’atteinte bulbaire) est également indispensable, de même qu’une évaluation par un ergothérapeute en cas de perte d’autonomie pour la mise en place d’aides techniques personnalisées.
Une prise en charge par un psychologue doit être proposée en cas de troubles anxiodépressifs, éventuellement associée à un traitement médicamenteux.
Le volet social ne doit pas être négligé
De même, une prise en charge par un service social est nécessaire afin d’informer les patients sur leurs droits et de les orienter pour la mise en place d’aides humaines et financières pouvant participer à la compensation du handicap.
Rôle des équipes de soins palliatifs
Parce que la prise en charge des patients atteints de SLA est complexe et nécessite des prises de décision parfois difficiles, que la maladie peut être responsable de symptômes altérant la qualité de vie (douleurs, encombrement respiratoire, troubles de l’humeur), l’appui d’une équipe de soins palliatifs devrait systématiquement être proposé au cours du suivi. Les objectifs sont d’optimiser le confort du patient, de discuter les stratégies thérapeutiques (introduction ou limitation et arrêt de certains traitements, notamment des techniques de suppléance vitale) et d’accompagner au mieux le patient sur les questions encadrant la fin de vie.
Que dire à vos patients ?
La SLA est une maladie neurodégénérative touchant les motoneurones, cellules nerveuses qui commandent les muscles volontaires permettant le contrôle des mouvements, de la parole, de la déglutition et de la respiration.
La cause de la SLA est inconnue, sauf dans de rares cas où la maladie peut être d’origine génétique (environ 10 %).
La maladie est évolutive mais avec une grande variabilité interindividuelle, et donc difficilement prévisible.
La prise en charge est multidisciplinaire, idéalement coordonnée par un centre SLA.
Le site de l’association pour la recherche sur la SLA et les autres maladies du motoneurone est une source fiable d’information : www.arsla.org
2. van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet 2017;390(10107):2084‑98.
3. Conférence de consensus HAS. Prise en charge des personnes atteintes de sclérose latérale amyotrophique – Recommandations professionnelles 2005.
4. Chiò A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler 2009;10(5‑6):310‑23.
5. Desport JC, Preux PM, Truong TC, Vallat JM, Sautereau D, Couratier P. Nutritional status is a prognostic factor for survival in ALS patients. Neurology 1999;53(5):1059‑63.
6. Lacomblez L, Bensimon G, Leigh PN, et al. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Lancet 1996;347(9013):1425-31.
7. Takei K, Tsuda K, Takahashi F, et al. Post-hoc analysis of open-label extension period of study MCI186-19 in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2017;18(sup1):64‑70.
8. Witzel S, Maier A, Steinbach R, et al. Safety and Effectiveness of Long-term Intravenous Administration of Edaravone for Treatment of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol 2022;79(2):121-30.
9. Paganoni S, Macklin EA, Hendrix S, et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N Engl J Med 2020;383(10):919‑30.
10. Miller TM, Cudkowicz ME, Genge A, et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med 2022;387(12):1099‑110.
Dans cet article
 Encadrés
Encadrés
La SLA est une pathologie neurodégénérative caractérisée par une atteinte progressive des motoneurones centraux et périphériques, responsable d’une paralysie progressive.
Son pronostic sévère conduit au décès, le plus souvent lié à une insuffisance respiratoire restrictive, dans des délais variables – en moyenne deux à cinq ans après l’apparition des premiers symptômes.
Le diagnostic repose en grande partie sur l’examen clinique, qui montre des signes d’atteinte des motoneurones. Un déficit moteur progressif amyotrophiant asymétrique d’un membre ou une dysarthrie associée à des troubles de la déglutition sont des tableaux cliniques classiques.
La prise en charge est multidisciplinaire et nécessite un suivi régulier, au mieux coordonné par un centre SLA, en lien avec le médecin traitant.
Les traitements restent largement dominés par les méthodes de suppléance respiratoire et nutritionnelle et par le traitement des symptômes, mais évoluent grâce à de nouvelles perspectives thérapeutiques.