Toujours y penser devant un anévrisme ou une dissection de l’aorte chez un sujet jeune, en l’absence d’hypertension artérielle et devant une ectopie du cristallin, car sa prise en charge maintenant standardisée a permis une augmentation de l’espérance de vie des patients de plus de 30 ans.
Le syndrome de Marfan est une maladie génétique autosomique dominante, généralement en rapport avec une mutation dans le gène de la fibrilline de type 1. C’est la plus fréquente des maladies génétiques responsables d’anévrisme de l’aorte ascendante. On estime qu’elle affecte environ 1/5 000 individus, soit une population d’environ 12 000 patients en France. Elle se caractérise par une combinaison variable d’atteintes cardiovasculaires, musculo-squelettiques, ophtalmologiques et pulmonaires qui témoignent d’une fragilité du tissu de soutien. Le pronostic vital est dominé par l’atteinte aortique (risque de dissection et de rupture), avec une espérance de vie spontanée de l’ordre de 40 ans dans les formes graves. Le pronostic fonctionnel dépend principalement des atteintes ophtalmologiques et musculo-squelettiques.
Critères diagnostiques
Le syndrome de Marfan est, à ce jour, défini par des critères essentiellement cliniques publiés en 1996 puis modifiés en 2009.1, 2 Ces critères sont complexes, témoignant de la difficulté diagnostique et illustrent la nécessaire collaboration de plusieurs spécialistes pour porter le diagnostic (v. tableau).
Les signes ou situations cliniques devant conduire à adresser les patients dans un centre expert ont été synthétisés dans un document du Réseau européen des maladies vasculaires rares.*
Cet article est centré sur le syndrome de Marfan, mais ces dernières années de nombreux gènes autres que celui de la fibrilline de type 1 ont été associés à des anévrismes de l’aorte ascendante, soit isolés (formes non syndromiques), soit associés à des signes cliniques (formes syndromiques). Les gènes associés peuvent se répartir en trois grandes familles :3
– les gènes codant des protéines de la matrice extracellulaire : le plus fréquent est le gène FBN1, qui code la fibrilline de type 1, microfibrille présente dans la matrice extracellulaire et responsable du syndrome de Marfan classique ; d’autres gènes ont été rapportés, qui sont en cause plus rarement (microfibril-associated protein 5 [MFAP5], filamin A LOX) ;
– les gènes codant l’appareil contractile des cellules musculaires lisses : gène codant l’actine (ACTA2), la myosine (MYH11) ou les protéines régulatrices de la contraction (myosin LC kinase [MYLCK], protein kinase G [PKG]). La physiopathologie proposée est que la cellule musculaire lisse ne perçoit pas normalement la tension appliquée à la paroi du fait de l’anomalie de la contraction cellulaire, et qu’elle va se transformer (de contractile, elle devient sécrétoire) pour permettre une destruction pour une meilleure reconstruction de la paroi aortique. Ce comportement est inadapté car déclenché par une erreur de perception et responsable de la fragilisation de la paroi aortique. Il s’agit généralement d’anévrismes isolés ;
– les gènes codant la voie du transforming growth factor bêta (TGF-bêta). Il s’agit d’une voie de signalisation qui indique à la cellule musculaire lisse de transcrire et traduire les gènes de reconstruction de la paroi aortique. Si ces gènes ne sont pas activés à bon escient, la réparation de la paroi est altérée, et un anévrisme se développe. Peuvent être en cause : les gènes codant le TGF-bêta2 ou 3 (souvent anévrismes isolés, pénétrance incomplète) ; les récepteurs du TGF-bêta, TGFBR1 et TGFBR2 (grande variabilité d’expression, avec parfois aucune traduction clinique [pénétrance incomplète], un anévrisme isolé, ou un syndrome malformatif, de Loeys-Dietz, associant hypertélorisme, luette bifide, rétrognatisme, tortuosité artérielle, les signes squelettiques du syndrome de Marfan classique et une maladie artérielle agressive), ou l’intermédiaire intracellulaire de la voie du TGF-bêta, SMAD3 (anévrismes isolés, arthrose précoce…).
Les signes ou situations cliniques devant conduire à adresser les patients dans un centre expert ont été synthétisés dans un document du Réseau européen des maladies vasculaires rares.*
Cet article est centré sur le syndrome de Marfan, mais ces dernières années de nombreux gènes autres que celui de la fibrilline de type 1 ont été associés à des anévrismes de l’aorte ascendante, soit isolés (formes non syndromiques), soit associés à des signes cliniques (formes syndromiques). Les gènes associés peuvent se répartir en trois grandes familles :3
– les gènes codant des protéines de la matrice extracellulaire : le plus fréquent est le gène FBN1, qui code la fibrilline de type 1, microfibrille présente dans la matrice extracellulaire et responsable du syndrome de Marfan classique ; d’autres gènes ont été rapportés, qui sont en cause plus rarement (microfibril-associated protein 5 [MFAP5], filamin A LOX) ;
– les gènes codant l’appareil contractile des cellules musculaires lisses : gène codant l’actine (ACTA2), la myosine (MYH11) ou les protéines régulatrices de la contraction (myosin LC kinase [MYLCK], protein kinase G [PKG]). La physiopathologie proposée est que la cellule musculaire lisse ne perçoit pas normalement la tension appliquée à la paroi du fait de l’anomalie de la contraction cellulaire, et qu’elle va se transformer (de contractile, elle devient sécrétoire) pour permettre une destruction pour une meilleure reconstruction de la paroi aortique. Ce comportement est inadapté car déclenché par une erreur de perception et responsable de la fragilisation de la paroi aortique. Il s’agit généralement d’anévrismes isolés ;
– les gènes codant la voie du transforming growth factor bêta (TGF-bêta). Il s’agit d’une voie de signalisation qui indique à la cellule musculaire lisse de transcrire et traduire les gènes de reconstruction de la paroi aortique. Si ces gènes ne sont pas activés à bon escient, la réparation de la paroi est altérée, et un anévrisme se développe. Peuvent être en cause : les gènes codant le TGF-bêta2 ou 3 (souvent anévrismes isolés, pénétrance incomplète) ; les récepteurs du TGF-bêta, TGFBR1 et TGFBR2 (grande variabilité d’expression, avec parfois aucune traduction clinique [pénétrance incomplète], un anévrisme isolé, ou un syndrome malformatif, de Loeys-Dietz, associant hypertélorisme, luette bifide, rétrognatisme, tortuosité artérielle, les signes squelettiques du syndrome de Marfan classique et une maladie artérielle agressive), ou l’intermédiaire intracellulaire de la voie du TGF-bêta, SMAD3 (anévrismes isolés, arthrose précoce…).
Atteinte cardiovasculaire
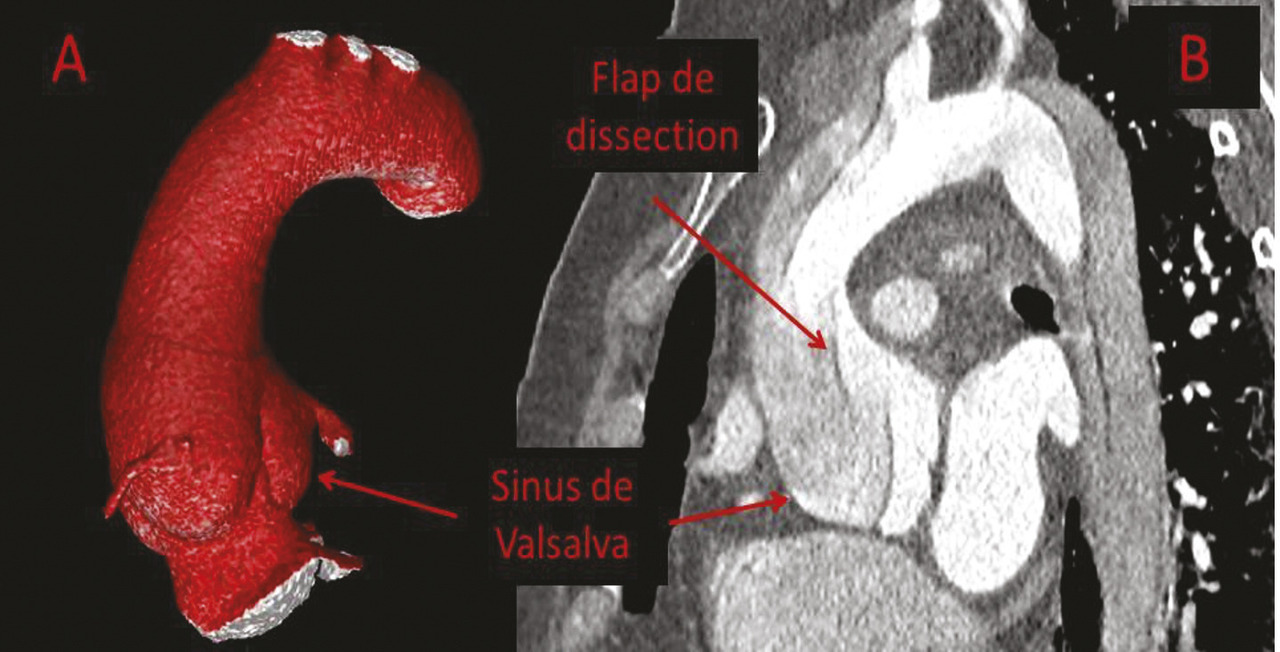
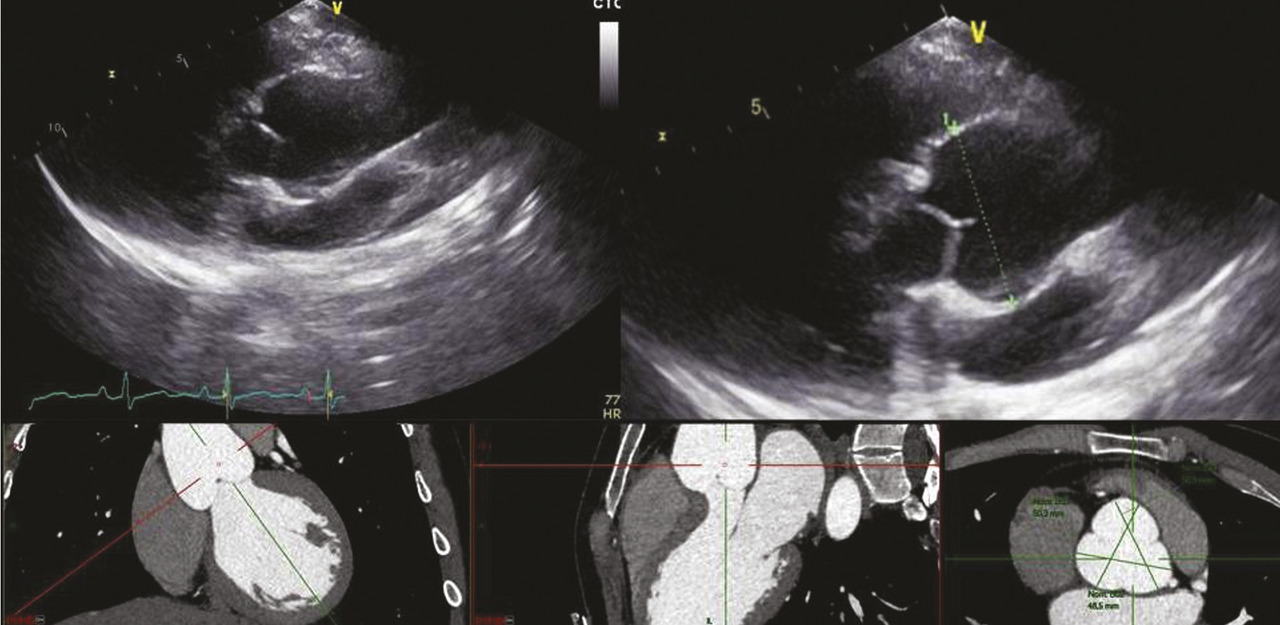
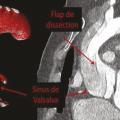
Sur le plan cardiovasculaire, le syndrome de Marfan se traduit principalement par une atteinte de la paroi aortique. La dilatation aortique prédomine au niveau des sinus de Valsalva dont le diamètre progresse en moyenne de 0,5 mm/an dans l’ensemble de la population Marfan adulte (mais chez certains patients la dilatation s’opère moins vite que chez d’autres). Le risque de dissection au niveau de l’aorte ascendante est corrélé au diamètre maximal au niveau des sinus de Valsalva (fig. 1 ). Cela justifie une surveillance des diamètres aortiques tous les ans, tout au long de la vie, même lorsque les diamètres sont dans les valeurs normales. Cette surveillance est généralement réalisée par échocardiographie et, chez les patients adultes, on confirme au moins une fois la fiabilité des mesures échographiques par une autre technique (généralement une tomodensitométrie injectée) [fig. 2 ].
Il est nécessaire de comparer le diamètre aortique obtenu aux valeurs normales théoriques en fonction de l’âge, du poids, du sexe et de la taille. Différents nomogrammes sont disponibles et doivent être utilisés en routine pour définir la normalité ou non des diamètres aortiques mesurés (calcul du Z-score**).
Si le risque principal de dissection aortique se situe sur l’aorte ascendante, l’atteinte aortique est diffuse, et il existe également un risque, plus faible, de dissection de l’aorte descendante.4 Cette dissection de l’aorte thoracique descendante peut survenir alors même que les diamètres de l’aorte ascendante et descendante sont normaux. Le risque de dissection de l’aorte descendante n’étant pas prévisible sur la base de critères cliniques ou radiologiques, une intervention prophylactique n’est pas possible.
L’atteinte d’artères périphériques autres que l’aorte est rare dans le syndrome de Marfan classique. Elle est moins rare dans les formes apparentées discutées plus haut, si bien qu’il est de règle dans ces formes non Marfan classique de faire une fois un bilan vasculaire complet des artères périphériques et cérébrales (dont la rentabilité est assez faible).
Enfin, sur le plan valvulaire, le prolapsus valvulaire mitral est fréquent, souvent bivalvulaire équilibré avec une fuite modérée qui nécessite rarement la chirurgie. Cependant, en cas de chirurgie mitrale, une plastie est préférée, même si elle peut être délicate du fait de l’étendue du prolapsus et de l’importance de la dilatation de l’anneau mitral.
Il est nécessaire de comparer le diamètre aortique obtenu aux valeurs normales théoriques en fonction de l’âge, du poids, du sexe et de la taille. Différents nomogrammes sont disponibles et doivent être utilisés en routine pour définir la normalité ou non des diamètres aortiques mesurés (calcul du Z-score**).
Si le risque principal de dissection aortique se situe sur l’aorte ascendante, l’atteinte aortique est diffuse, et il existe également un risque, plus faible, de dissection de l’aorte descendante.4 Cette dissection de l’aorte thoracique descendante peut survenir alors même que les diamètres de l’aorte ascendante et descendante sont normaux. Le risque de dissection de l’aorte descendante n’étant pas prévisible sur la base de critères cliniques ou radiologiques, une intervention prophylactique n’est pas possible.
L’atteinte d’artères périphériques autres que l’aorte est rare dans le syndrome de Marfan classique. Elle est moins rare dans les formes apparentées discutées plus haut, si bien qu’il est de règle dans ces formes non Marfan classique de faire une fois un bilan vasculaire complet des artères périphériques et cérébrales (dont la rentabilité est assez faible).
Enfin, sur le plan valvulaire, le prolapsus valvulaire mitral est fréquent, souvent bivalvulaire équilibré avec une fuite modérée qui nécessite rarement la chirurgie. Cependant, en cas de chirurgie mitrale, une plastie est préférée, même si elle peut être délicate du fait de l’étendue du prolapsus et de l’importance de la dilatation de l’anneau mitral.
Atteinte musculo-squelettique
Les patients atteints du syndrome de Marfan ont souvent un morphotype longiligne caractéristique, avec une grande taille. Ils peuvent aussi avoir des anomalies caractéristiques au niveau du crâne et du visage, avec une hypoplasie malaire, une dolichocéphalie, un rétrognatisme et une enophtalmie.
La croissance excessive des os longs peut entraîner un pectus excavatum ou recurvatum, une arachnodactylie, et/ou une scoliose. Les patients peuvent également avoir les pieds plats. Les conséquences sont essentiellement fonctionnelles, avec des douleurs parfois difficiles à soulager, des problèmes esthétiques d’image qui peuvent amener les patients à recourir à la chirurgie esthétique. La scoliose et les pieds plats nécessitent souvent une prise en charge orthopédique (orthèse, corset, chirurgie).
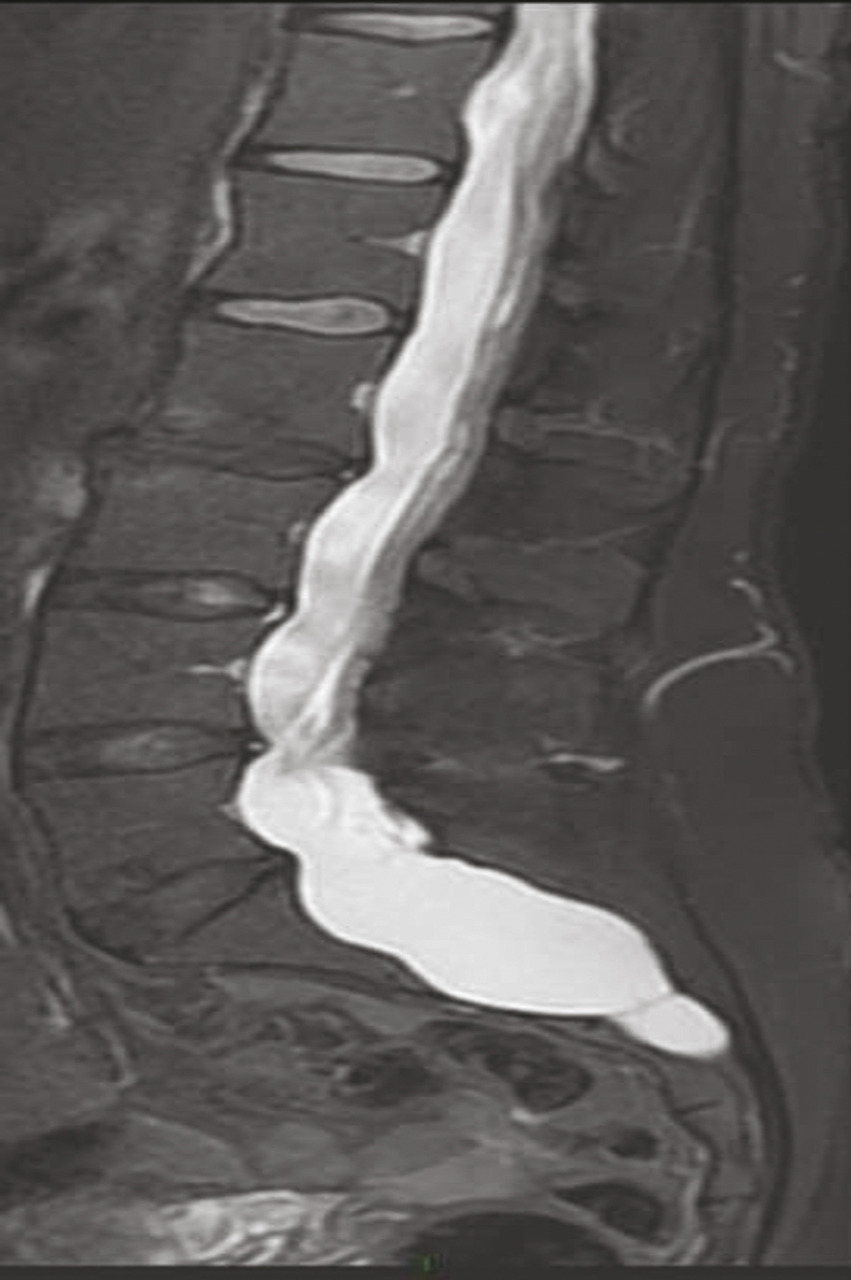
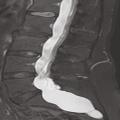
Les vertèbres peuvent être déformées par une ectasie du sac dural (fig. 3 ), visible essentiellement à la tomodensitométrie ou à l’imagerie par résonance magnétique (IRM). Les symptômes associés à cette ectasie durale restent mal connus. Elle peut être associée à des céphalées en rapport avec une hypotension du liquide céphalorachidien dont la symptomatologie est proche du syndrome post-ponction lombaire.
La croissance excessive des os longs peut entraîner un pectus excavatum ou recurvatum, une arachnodactylie, et/ou une scoliose. Les patients peuvent également avoir les pieds plats. Les conséquences sont essentiellement fonctionnelles, avec des douleurs parfois difficiles à soulager, des problèmes esthétiques d’image qui peuvent amener les patients à recourir à la chirurgie esthétique. La scoliose et les pieds plats nécessitent souvent une prise en charge orthopédique (orthèse, corset, chirurgie).
Les vertèbres peuvent être déformées par une ectasie du sac dural (
Atteinte ophtalmologique
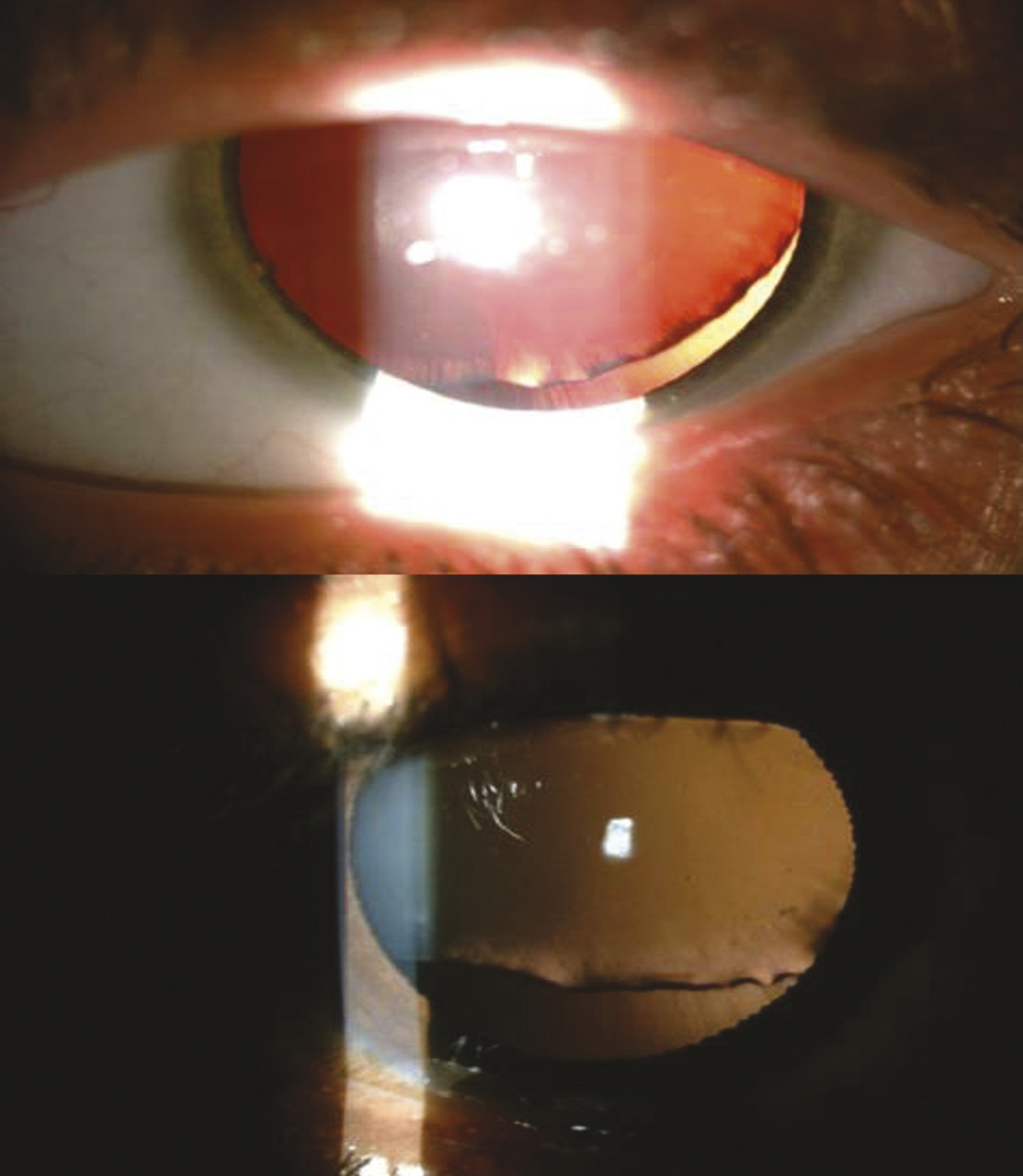
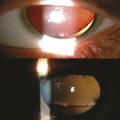
L’ectopie du cristallin est un signe diagnostique majeur (comme l’anévrisme aortique), elle est souvent supérieure et temporale, et souvent incomplète (fig. 4 ). Lorsqu’elle est importante, elle peut conduire à la cécité. Son diagnostic justifie une dilatation pupillaire soigneuse et un examen à la lampe à fente. Les autres signes ophtalmologiques sont les cornées plates et la myopie secondaire à une longueur axiale augmentée (la chirurgie de la myopie n’est souvent pas indiquée).
Autres atteintes
Une atteinte pulmonaire peut également être observée, avec un pneumothorax, qui est en fait assez rare dans cette population.
L’atteinte cutanée est fréquente, avec la présence de vergetures sur tout le corps, la localisation sur le devant des épaules étant plus spécifique du syndrome de Marfan.
L’atteinte cutanée est fréquente, avec la présence de vergetures sur tout le corps, la localisation sur le devant des épaules étant plus spécifique du syndrome de Marfan.
Traitement
Sur le plan cardiovasculaire, l’objectif est d’éviter la survenue d’une dissection aortique. La prise en charge comporte donc des recommandations d’hygiène de vie, un traitement médicamenteux et une intervention prophylactique de remplacement de l’aorte ascendante.
On recommande d’éviter les efforts physiques lors desquels la pression artérielle s’élève trop ou trop brutalement : les efforts isométriques (tels que l’haltérophilie, la musculation) sont à éviter ainsi que les sports dynamiques à haute intensité (basket, badminton, handball, football…). La compétition est contre-indiquée pour la plupart des sports. Les efforts d’endurance, par exemple la natation, la course à pied, le vélo hors compétition sont en revanche conseillés. On s’appuie volontiers sur la classification américaine des sports en fonction de leur composante dynamique et statique.5
Le traitement médical repose sur les bêtabloquants, dont la démonstration de l’efficacité a été faite sur le ralentissement de la dilatation aortique dans une étude ancienne randomisée et contre placebo en simple insu, réalisée par l’équipe de l’hôpital Johns-Hopkins, chez 70 patients de plus de 12 ans.6 Cela justifie donc de proposer le traitement bêtabloquant quel que soit le diamètre de l’aorte ascendante en cas de diagnostic de syndrome de Marfan. La molécule généralement utilisée est le bisoprolol à la dose de 10 mg si possible. En cas de contre-indication aux bêtabloquants (asthme), on propose un traitement par inhibiteurs calciques dont les propriétés hémodynamiques sont similaires (bradycardie et inotropisme négatifs) dans l’idée de limiter le stress appliqué à la paroi aortique, et donc le risque de dilatation et de dissection. Les espoirs mis dans le losartan, issus de travaux réalisés chez la souris,7 n’ont pas été confirmés chez l’homme après plusieurs études randomisées.
Enfin, une chirurgie prophylactique de remplacement de la racine de l’aorte est préconisée en fonction du diamètre maximal aortique et du gène muté.8 Le risque de dissection augmente lorsque le diamètre maximal aortique atteint 50 mm chez les adultes ayant une mutation dans le gène FBN1, et le risque opératoire devient alors plus faible que le risque annuel spontané de dissection aortique.9 Les dernières recommandations européennes de 2017 proposent donc une intervention chirurgicale lorsque le diamètre des sinus de Valsalva atteint 50 mm.8 Les techniques chirurgicales de préservation de la valve aortique (techniques de Tirone-David ou de Lansac) doivent être la règle, ce qui implique de choisir une équipe chirurgicale qui maîtrise cette technique. En aucun cas, du tissu aortique natif ne doit être laissé en place.
De même, le traitement médical et les règles de vie sont à poursuivre après remplacement de l’aorte ascendante.
On recommande d’éviter les efforts physiques lors desquels la pression artérielle s’élève trop ou trop brutalement : les efforts isométriques (tels que l’haltérophilie, la musculation) sont à éviter ainsi que les sports dynamiques à haute intensité (basket, badminton, handball, football…). La compétition est contre-indiquée pour la plupart des sports. Les efforts d’endurance, par exemple la natation, la course à pied, le vélo hors compétition sont en revanche conseillés. On s’appuie volontiers sur la classification américaine des sports en fonction de leur composante dynamique et statique.5
Le traitement médical repose sur les bêtabloquants, dont la démonstration de l’efficacité a été faite sur le ralentissement de la dilatation aortique dans une étude ancienne randomisée et contre placebo en simple insu, réalisée par l’équipe de l’hôpital Johns-Hopkins, chez 70 patients de plus de 12 ans.6 Cela justifie donc de proposer le traitement bêtabloquant quel que soit le diamètre de l’aorte ascendante en cas de diagnostic de syndrome de Marfan. La molécule généralement utilisée est le bisoprolol à la dose de 10 mg si possible. En cas de contre-indication aux bêtabloquants (asthme), on propose un traitement par inhibiteurs calciques dont les propriétés hémodynamiques sont similaires (bradycardie et inotropisme négatifs) dans l’idée de limiter le stress appliqué à la paroi aortique, et donc le risque de dilatation et de dissection. Les espoirs mis dans le losartan, issus de travaux réalisés chez la souris,7 n’ont pas été confirmés chez l’homme après plusieurs études randomisées.
Enfin, une chirurgie prophylactique de remplacement de la racine de l’aorte est préconisée en fonction du diamètre maximal aortique et du gène muté.8 Le risque de dissection augmente lorsque le diamètre maximal aortique atteint 50 mm chez les adultes ayant une mutation dans le gène FBN1, et le risque opératoire devient alors plus faible que le risque annuel spontané de dissection aortique.9 Les dernières recommandations européennes de 2017 proposent donc une intervention chirurgicale lorsque le diamètre des sinus de Valsalva atteint 50 mm.8 Les techniques chirurgicales de préservation de la valve aortique (techniques de Tirone-David ou de Lansac) doivent être la règle, ce qui implique de choisir une équipe chirurgicale qui maîtrise cette technique. En aucun cas, du tissu aortique natif ne doit être laissé en place.
De même, le traitement médical et les règles de vie sont à poursuivre après remplacement de l’aorte ascendante.
Surveillance
Une surveillance annuelle par échocardiographie est recommandée quels que soient le diamètre maximal aortique ou les antécédents de chirurgie aortique.
Après dissection de l’aorte descendante, une tomodensitométrie aortique synchronisée à l’électrocardiogramme (ECG) annuel est indiquée.
En cas de chirurgie aortique, une tomodensitométrie est recommandée avant l’intervention, dans les 3 à 6 mois postopératoires, puis tous les 5 ans pour juger de la qualité du montage et surveiller l’évolution du reste de l’aorte thoraco-abdominale.
Après dissection de l’aorte descendante, une tomodensitométrie aortique synchronisée à l’électrocardiogramme (ECG) annuel est indiquée.
En cas de chirurgie aortique, une tomodensitométrie est recommandée avant l’intervention, dans les 3 à 6 mois postopératoires, puis tous les 5 ans pour juger de la qualité du montage et surveiller l’évolution du reste de l’aorte thoraco-abdominale.
Une grossesse à risque
Chez les femmes atteintes d’un syndrome de Marfan, la grossesse est associée à un sur-risque de dissection aortique dont il faut informer les femmes en âge de procréer. Il est recommandé de réaliser une évaluation préconceptionnelle, dans un centre de référence ou de compétences avant la grossesse. On conseille l’accouchement dans une maternité qui a l’habitude de prendre en charge des patientes atteintes d’un syndrome de Marfan, en relation avec un service de cardiologie et si possible avec la chirurgie cardiaque accessible. Se pose parfois le problème de l’épidurale chez des patientes ayant une scoliose ou/et une ectasie durale.
La prise en charge de la grossesse pose des problèmes particuliers : schématiquement, le risque de dissection de l’aorte ascendante est considéré comme faible lorsque le diamètre maximal aortique au niveau des sinus de Valsalva est inférieur à 40 mm. On autorise alors un accouchement par voie basse, sous bêtabloquants, dans une maternité qui a l’habitude de prendre en charge des patientes atteintes d’un syndrome de Marfan.
Lorsque le diamètre maximal aortique est supérieur à 45 mm, le risque de dissection de l’aorte ascendante contre-indique la grossesse, et la chirurgie prophylactique aortique peut être proposée en vue de mener une grossesse.10
Lorsque le diamètre maximal aortique est entre 40 et 45 mm, la décision est prise au cas par cas, en fonction du gène muté, de l’histoire familiale et de l’évolution des diamètres aortiques au cours des dernières années.
Une surveillance échocardiographique étroite doit être mise en place avec une échographie à 3 mois, 6 mois, puis tous les mois au cours du troisième trimestre et au décours de l’accouchement.
Enfin, le traitement par bêtabloquants doit être poursuivi tout au long de la grossesse et en post-partum, ce qui contre-indique l’allaitement.
La prise en charge de la grossesse pose des problèmes particuliers : schématiquement, le risque de dissection de l’aorte ascendante est considéré comme faible lorsque le diamètre maximal aortique au niveau des sinus de Valsalva est inférieur à 40 mm. On autorise alors un accouchement par voie basse, sous bêtabloquants, dans une maternité qui a l’habitude de prendre en charge des patientes atteintes d’un syndrome de Marfan.
Lorsque le diamètre maximal aortique est supérieur à 45 mm, le risque de dissection de l’aorte ascendante contre-indique la grossesse, et la chirurgie prophylactique aortique peut être proposée en vue de mener une grossesse.10
Lorsque le diamètre maximal aortique est entre 40 et 45 mm, la décision est prise au cas par cas, en fonction du gène muté, de l’histoire familiale et de l’évolution des diamètres aortiques au cours des dernières années.
Une surveillance échocardiographique étroite doit être mise en place avec une échographie à 3 mois, 6 mois, puis tous les mois au cours du troisième trimestre et au décours de l’accouchement.
Enfin, le traitement par bêtabloquants doit être poursuivi tout au long de la grossesse et en post-partum, ce qui contre-indique l’allaitement.
Conseil génétique et enquête familiale
La transmission de la maladie se fait selon le mode dominant autosomique : un patient atteint a un risque sur deux de transmettre à son enfant la mutation pathogène dans le gène FBN1 sans préférence de sexe. Un des deux parents est atteint sauf si la mutation apparaît pour la première fois (néomutation), ce qui est le cas chez 25 % des patients.
Cela a deux conséquences :
– il faut rechercher un anévrisme aortique chez les apparentés du premier degré avant qu’une dissection ne survienne. Cela justifie de rechercher la présence des signes cliniques et échocardiographiques chez les apparentés d’un patient atteint ;
– il est possible de réaliser une étude de biologie moléculaire à la recherche de la mutation causale. Elle permet de confirmer le diagnostic dans les formes douteuses et facilite l’enquête familiale. Elle reste néanmoins soumise au filtre du laboratoire de biologie moléculaire, du fait de sa difficulté (le gène est long et chaque famille a une mutation qui lui est propre) et du faible rendement de la recherche de mutation en l’absence de signes cliniques suffisants (parce que beaucoup d’anévrismes ne sont pas d’origine purement génétique). En pratique, on demande pour que la recherche de biologie moléculaire soit effectuée qu’il y ait11, 12 soit une atteinte nette de deux systèmes ; soit que la forme familiale soit attestée ; soit que l’anévrisme survienne chez un sujet jeune (< 45 ans).
Cela a deux conséquences :
– il faut rechercher un anévrisme aortique chez les apparentés du premier degré avant qu’une dissection ne survienne. Cela justifie de rechercher la présence des signes cliniques et échocardiographiques chez les apparentés d’un patient atteint ;
– il est possible de réaliser une étude de biologie moléculaire à la recherche de la mutation causale. Elle permet de confirmer le diagnostic dans les formes douteuses et facilite l’enquête familiale. Elle reste néanmoins soumise au filtre du laboratoire de biologie moléculaire, du fait de sa difficulté (le gène est long et chaque famille a une mutation qui lui est propre) et du faible rendement de la recherche de mutation en l’absence de signes cliniques suffisants (parce que beaucoup d’anévrismes ne sont pas d’origine purement génétique). En pratique, on demande pour que la recherche de biologie moléculaire soit effectuée qu’il y ait11, 12 soit une atteinte nette de deux systèmes ; soit que la forme familiale soit attestée ; soit que l’anévrisme survienne chez un sujet jeune (< 45 ans).
Pronostic
La compréhension de la physiopathologie du syndrome de Marfan et la prise en charge des patients ont beaucoup évolué ces dernières années, ce qui a modifié le pronostic. Depuis la standardisation de sa prise en charge, l’espérance de vie des patients a augmenté de plus de 30 ans. Antérieurement, 80 % des patients mouraient des conséquences de la dilatation aortique (dissection ou fuite aortique avec insuffisance cardiaque), et la moitié des patients ayant une forme grave décédaient avant l’âge de 40 ans. La survenue d’une dissection de l’aorte thoracique descendante est plus rare mais reste imprévisible.
Conclusion
Le syndrome de Marfan est la pathologie aortique d’origine génétique la plus fréquente.
La multiplicité des signes cliniques et des systèmes atteints souligne l’importance de l’expertise d’un centre de référence ou de compétences pour porter le diagnostic. La connaissance du diagnostic permet, avec la prise en charge actuelle, de transformer le pronostic des patients. L’évocation du diagnostic doit donc être systématique devant tout anévrisme ou dissection de l’aorte chez un sujet jeune en l’absence d’hypertension artérielle et devant une ectopie du cristallin. Enfin, une enquête familiale est indispensable en cas de diagnostic de syndrome de Marfan ou en cas d’anévrisme aortique ou de dissection aortique.
La multiplicité des signes cliniques et des systèmes atteints souligne l’importance de l’expertise d’un centre de référence ou de compétences pour porter le diagnostic. La connaissance du diagnostic permet, avec la prise en charge actuelle, de transformer le pronostic des patients. L’évocation du diagnostic doit donc être systématique devant tout anévrisme ou dissection de l’aorte chez un sujet jeune en l’absence d’hypertension artérielle et devant une ectopie du cristallin. Enfin, une enquête familiale est indispensable en cas de diagnostic de syndrome de Marfan ou en cas d’anévrisme aortique ou de dissection aortique.
Références
1. De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996;62:417‑26.
2. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476‑85.
3. Michel JB, Jondeau G, Milewicz DM. From genetics to response to injury: vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc Res 2018;114:578‑89.
4. Mimoun L, Detaint D, Hamroun D, Arnoult F, Delorme G, Gautier M, et al. Dissection in Marfan syndrome: the importance of the descending aorta. Eur Heart J 2011;32:443‑9.
5. Levine BD, Baggish AL, Kovacs RJ, Link MS, Maron MS, Mitchell JH. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 1: classification of sports: dynamic, static, and impact: a scientific statement from the American Heart Association and American College of Cardiology. J Am Coll Cardiol 2015;66:2350‑5.
6. Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med 1994;330:1335‑41.
7. Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006;312:117‑21.
8. Baumgartner H, Falk V, Bax JJ, et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur Heart J 2017;38:2739‑91.
9. Milleron O, Arnoult F, Delorme G, et al. Pathogenic FBN1 genetic variation and aortic dissection in patients with Marfan syndrome. J Am Coll Cardiol 2020;75:843‑53.
10. Erbel R, Aboyans V, Boileau C, et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The task force for the diagnosis and treatment of aortic diseases of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2873‑926.
11. Stheneur C, Collod-Béroud G, Faivre L, et al. Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. Eur J Hum Genet 2009;17:1121‑8.
12. Arnaud P, Hanna N, Benarroch L, et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet Med 2019;21:2015-24.
2. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476‑85.
3. Michel JB, Jondeau G, Milewicz DM. From genetics to response to injury: vascular smooth muscle cells in aneurysms and dissections of the ascending aorta. Cardiovasc Res 2018;114:578‑89.
4. Mimoun L, Detaint D, Hamroun D, Arnoult F, Delorme G, Gautier M, et al. Dissection in Marfan syndrome: the importance of the descending aorta. Eur Heart J 2011;32:443‑9.
5. Levine BD, Baggish AL, Kovacs RJ, Link MS, Maron MS, Mitchell JH. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: task force 1: classification of sports: dynamic, static, and impact: a scientific statement from the American Heart Association and American College of Cardiology. J Am Coll Cardiol 2015;66:2350‑5.
6. Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med 1994;330:1335‑41.
7. Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006;312:117‑21.
8. Baumgartner H, Falk V, Bax JJ, et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur Heart J 2017;38:2739‑91.
9. Milleron O, Arnoult F, Delorme G, et al. Pathogenic FBN1 genetic variation and aortic dissection in patients with Marfan syndrome. J Am Coll Cardiol 2020;75:843‑53.
10. Erbel R, Aboyans V, Boileau C, et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The task force for the diagnosis and treatment of aortic diseases of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2873‑926.
11. Stheneur C, Collod-Béroud G, Faivre L, et al. Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. Eur J Hum Genet 2009;17:1121‑8.
12. Arnaud P, Hanna N, Benarroch L, et al. Genetic diversity and pathogenic variants as possible predictors of severity in a French sample of nonsyndromic heritable thoracic aortic aneurysms and dissections (nshTAAD). Genet Med 2019;21:2015-24.
Dans cet article


Résumé
Le syndrome de Marfan est une maladie génétique dominante autosomique qui touche environ 1 personne sur 5 000. Elle se traduit par une fragilité de la paroi aortique (avec dilatation progressive et risque de dissection), un prolapsus valvulaire mitral, des signes ophtalmologiques (ectopie du cristallin, cornées plates, myopie forte), des signes squelettiques (grande taille, déformation thoracique avec pectus, scoliose, arachnodactylie, pieds plats), des signes cutanés (vergetures, surtout à l’avant des épaules) et une ectasie durale. Le gène en cause est généralement celui codant la fibrilline 1. Le traitement repose sur les bêtabloquants, la contre-indication aux sports violents, et la chirurgie aortique prophylactique lorsque le diamètre maximal de l’aorte (au niveau des sinus de Valsalva) dépasse 50 mm. De nombreux syndromes apparentés ont été découverts ces dernières années.