Pour les patients atteints d’un syndrome d’Ehlers-Danlos, c’est souvent des années d’errance diagnostique, tellement la symptomatologie clinique est variée. Et bien souvent ce n’est qu’à un stade avancé de la maladie, où le handicap s’est installé, que le diagnostic est enfin posé.
Un protocole national de diagnostic et de soins vient d’être mis en place, afin de mieux informer les personnels soignants sur cette pathologie, et optimiser la prise en charge et le suivi des patients, en collaboration avec les centres de référence.
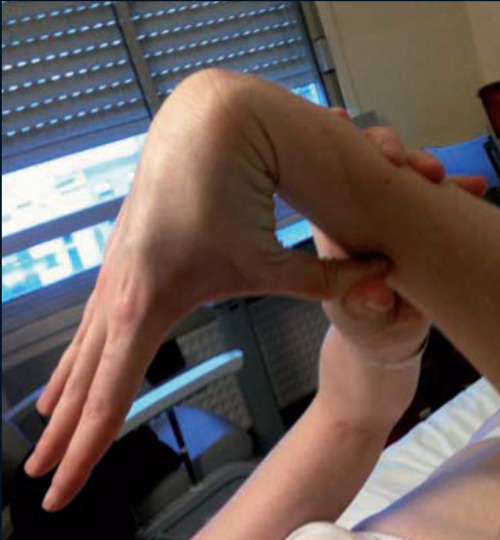
Les syndromes d’Ehlers-Danlos sont un groupe hétérogène de maladies héréditaires du tissu conjonctif caractérisées par la triade : hyperlaxité articulaire, hyperélasticité cutanée et fragilité des tissus conjonctifs. Ils sont dus à des anomalies de biosynthèse et/ou de structure de protéines de la matrice extracellulaire. Leur prévalence est estimée à 1 pour 5 000. La classification de New York 2017 décrit 13 types de syndrome d’Ehlers-Danlos. Ces différents types n’exposent pas aux mêmes complications et leur pronostic est donc différent. Le diagnostic final repose sur la confirmation moléculaire du diagnostic.
Une fois le diagnostic posé, une prise en charge spécialisée est nécessaire, face aux nombreuses comorbidités qui caractérisent ces syndromes ; sans négliger la prise en charge de la douleur parfois banalisée à tort, chez ces patients !
Alexandra Karsenty, La Revue du Praticien
En savoir plus :
Vivre avec... Le syndrome d’Ehlers Danlos
Hyperlaxité articulaire : quand évoquer un syndrome d’Ehlers-Danlos ?