Cette maladie clonale affecte la cellule souche hématopoïétique et survient essentiellement chez le sujet âgé. Elle se caractérise par des anomalies morphologiques et une hématopoïèse (fabrication des cellules sanguines) inefficace .
Le terme myélodysplasie correspond à une définition cytologique traduisant une anomalie qualitative des cellules composant la moelle osseuse observée au microscope. En pratique, on parle plutôt de dysplasie médullaire. Le syndrome myélodysplasique correspond à la maladie hématologique résultant de ces dysfonctionnements.
Les anomalies biologiques sous-jacentes sont en rapport avec des modifications cytogénétiques (aberrations chromosomiques), génomiques (mutations acquises) et épigénétiques (essentiellement méthylation de l’ADN) des cellules de la moelle osseuse devenues clonales.1 Le micro-environnement médullaire joue également un rôle.
C’est une maladie très hétérogène dans sa présentation clinique et dans son pronostic. On distingue ainsi classiquement les syndromes myélodysplasiques (SMD) de « faible risque » dont les symptômes sont liés essentiellement aux cytopénies (anémie, neutropénie et thrombopénie) et les SMD de « haut risque » dont le problème clinique réside dans le risque de transformation en leucémie aiguë myéloblastique (LAM). La progression vers une LAM est observée chez environ un tiers des patients, du fait d’une évolution de la susceptibilité des progéniteurs médullaires de l’apoptose vers la prolifération.
Une maladie du sujet âgé
Le SMD est l’hémopathie la plus fréquente du sujet âgé, avec une incidence globale d’environ 4 cas pour 100 000 habitants et par an et pouvant atteindre 70 cas pour 100 000 habitants et par an après 70 ans.2
La médiane d’âge se situe entre 65 et 70 ans, avec une légère prédominance chez les hommes. Seuls 8 à 10 % des patients ont moins de 50 ans, et les formes de l’enfant sont très rares, souvent associées à un contexte familial.
Avec le vieillissement de la population, le nombre de personnes atteintes de SMD pourrait continuer de croître ces prochaines années, d’autant que l’évolution des connaissances à propos de cette maladie concourt à réaliser de plus en plus d’explorations à sa recherche.
Le vieillissement en cause
Dans 80 % des situations, aucune autre étiologie spécifique n’est retrouvée.
Cependant, certaines anomalies génétiques constitutionnelles ainsi que les traitements anticancéreux (chimio-, radiothérapie) ou encore certains facteurs environnementaux favorisent la survenue des SMD.
Prédisposition génétique
On estime entre 5 et 10 % le nombre de SMD en rapport avec un terrain génétique. Toutefois, l’identification récente de nouveaux gènes de prédisposition aux hémopathies myéloïdes laisse penser que leur fréquence est sous-estimée.
Ces causes génétiques résultent parfois de l’évolution de maladies syndromiques pédiatriques (trisomie 21, maladie de Fanconi, téloméropathies) ou de prédispositions à la survenue d’hémopathies à l’âge adulte en rapport avec des variants constitutionnels patho- géniques (GATA2, RUNX1, DDX41).
Exposition aux agents anticancéreux
Les progrès dans la prise en charge des cancers sont associés à l’observation plus fréquente d’hémopathies induites par l’effet mutagène des cytotoxiques (chimiothérapies de type alkylants, analogues de purine ou inhibiteurs de la topo-isomérase II) et des radiothérapies. Les SMD secondaires à l’exposition aux anticancéreux représentent 10 à 15 % des cas et sont plutôt de moins bon pronostic.
Facteurs de risque environnementaux
Certaines expositions, y compris professionnelles, contribuent à la survenue de ce syndrome. Les agents chimiques (benzène, solvants), les radiations ionisantes ou encore le tabagisme actif peuvent être incriminés.
Quand évoquer un SMD ?
Les anomalies de l’hémogramme
Les anomalies de l’hémogramme (ou numération formule sanguine [NFS]) sont le plus souvent à l’origine de la suspicion d’un SMD.
Toute cytopénie d’origine centrale doit faire évoquer une maladie de la moelle osseuse, dont un SMD. D’installation progressive, la plus fréquente (80 % des cas) est l’anémie. Elle est volontiers normo- ou macrocytaire, normochrome et non régénérative. La thrombopénie et la neutropénie sont couramment associées à l’anémie, plus rarement isolées ; elles doivent aussi faire suspecter ce syndrome. Enfin, un pourcentage de monocytes circulants supérieur à 10 % fait suggérer une leucémie myélomonocytaire chronique (LMMC), forme frontière avec les syndromes myéloprolifératifs (SMP) [
Les anomalies de l’hémogramme sont souvent de découverte fortuite ; c’est un examen fréquent dans la surveillance systématique des patients âgés. Parfois, elles sont révélées par des symptômes liés aux conséquences des cytopénies.
Des signes cliniques en rapport avec les cytopénies
Bien que ces derniers soient les plus fréquents, une altération de l’état général est également possible.
L’anémie se manifeste par un essoufflement à l’effort, une asthénie ou des symptômes cardiaques majorés chez les patients avec des comorbidités cardiopulmonaires. L’examen clinique objective de manière inconstante une pâleur cutanéo-conjonctivale ou une tachycardie. L’installation de cette anémie est progressive, les signes cliniques graves sont donc souvent absents. Toutefois, il est recommandé de faire un électrocardiogramme, à la recherche de souffrance myocardique chez les sujets avec une anémie profonde, surtout dans un contexte de cardiopathie connue.
La thrombopénie se déclare occasionnellement par un syndrome hémorragique (épistaxis, gingivorragie ; parfois plus sévère : digestif, cérébral). Une thombopathie y est possiblement associée.
La neutropénie expose les patients à un risque infectieux, essentiellement bactérien. Rarement inaugurales, les complications infectieuses sont susceptibles de mettre en jeu le pronostic vital du patient.
Formes atypiques
Certains syndromes spécifiques (
– la splénomégalie peut être présente dans les formes classiques, plus volontiers dans les syndromes frontières SMD/SMP, ou dans la LMMC ;
– des maladies inflammatoires ou auto-immunes (vascularites, connectivites, atteintes rhumatologiques inflammatoires), parfois symptomatiques, amènent quelquefois à découvrir un SMD ;
– les localisations extrahématologiques sont très rares. Une des plus régulièrement observées est l’atteinte cutanée. Elle peut traduire un syndrome de Sweet (polynucléaires neutrophiles), une localisation blastique (plus proche de la LAM) ou une localisation spécifique de SMD.
Comment faire le diagnostic ?
L’examen clé est le myélogramme. Il permet d’affirmer le diagnostic tandis que la cytogénétique vient affiner le pronostic et la décision thérapeutique. La biologie moléculaire est souvent réalisée dans les centres spécialisés, avec une place grandissante dans la réflexion diagnostique, pronostique et thérapeutique.
Myélogramme
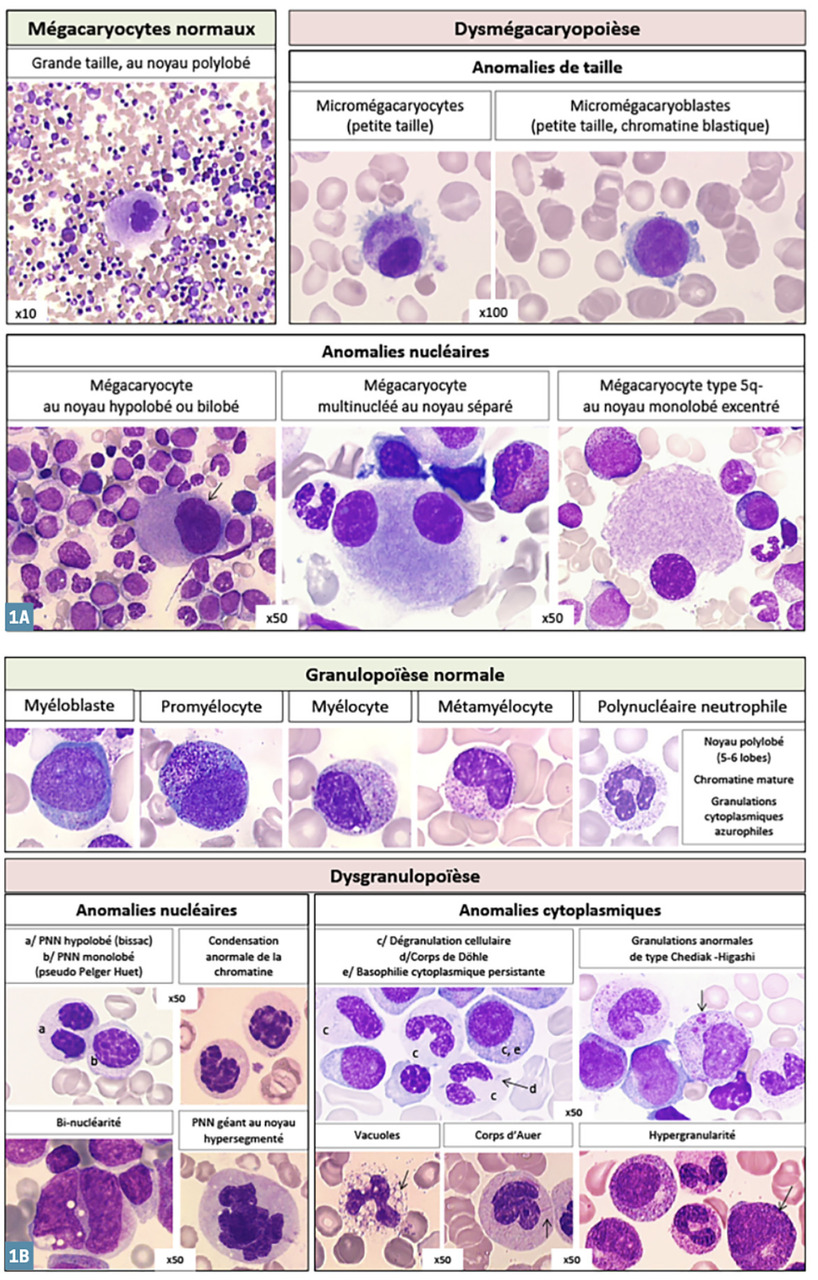
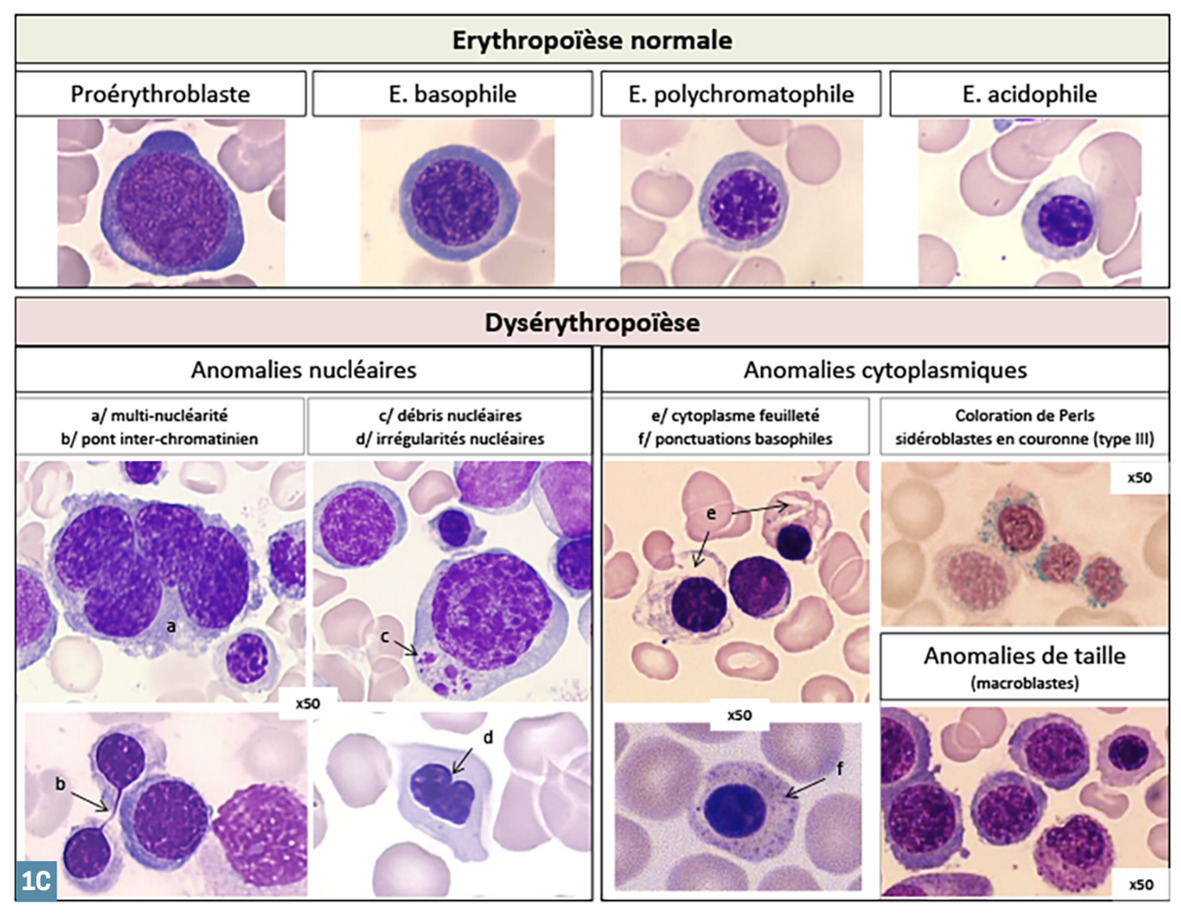
Indispensable au diagnostic, il est obtenu par ponction/aspiration de la moelle osseuse au niveau de l’os sternal ou iliaque. L’étalement sur lame et coloration autorise ensuite l’analyse morphologique au microscope, déterminante pour affirmer ou non un syndrome myélodysplasique. En effet, on observe des anomalies morphologiques (dysplasies) sur une ou plusieurs des 3 lignées (rouge, blanche, plaquettes) (
Selon la classification OMS, on parle de dysplasie d’une lignée lorsqu’au moins 10 % des cellules de cette lignée sont morphologiquement anormales.
La richesse de la moelle est variable, mais le plus souvent normale ou augmentée. Dans certains cas, elle est pauvre (cf. diagnostics différentiels).
À noter que certains SMD induisent des signes très modérés de dysplasie malgré des cytopénies signifiantes, et que le diagnostic est susceptible d’être porté ou éliminé en confrontant ces résultats à ceux de cytogénétique et parfois de biologie moléculaire.
C’est lors du myélogramme que sont également prélevées les cellules médullaires nécessaires au caryotype et à la biologie moléculaire.
Cytogénétique
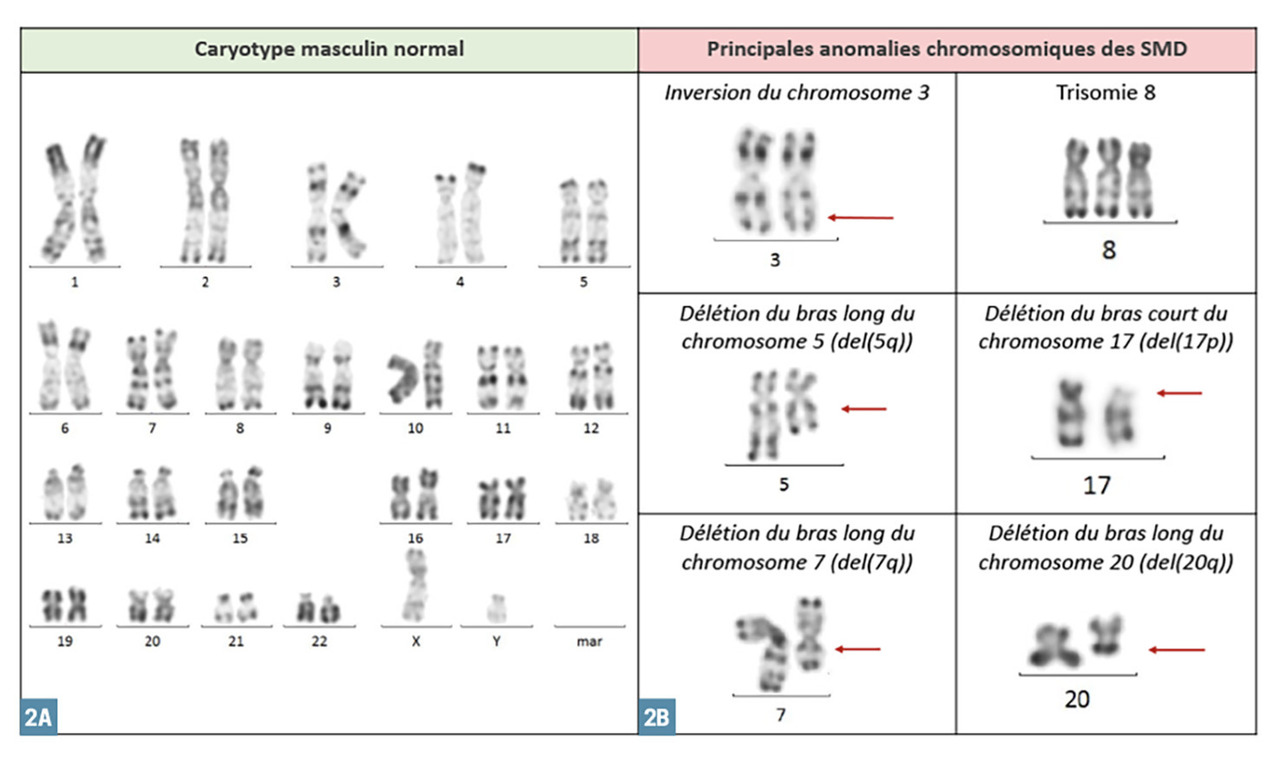
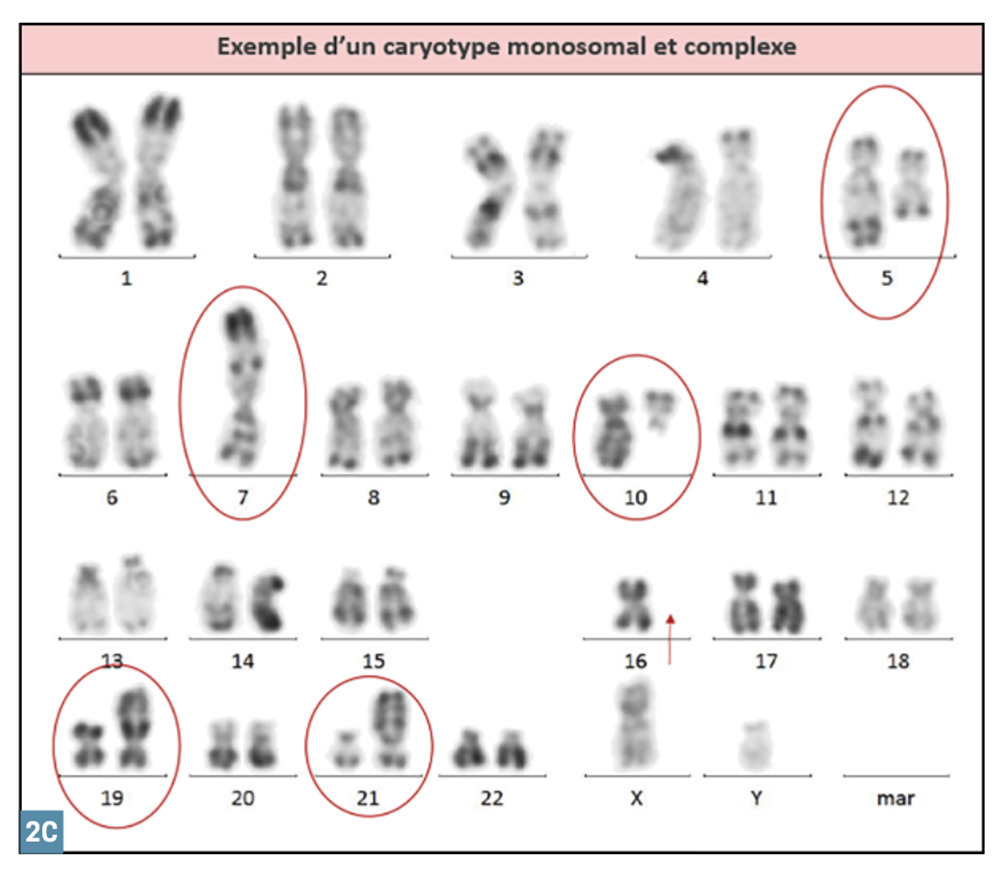
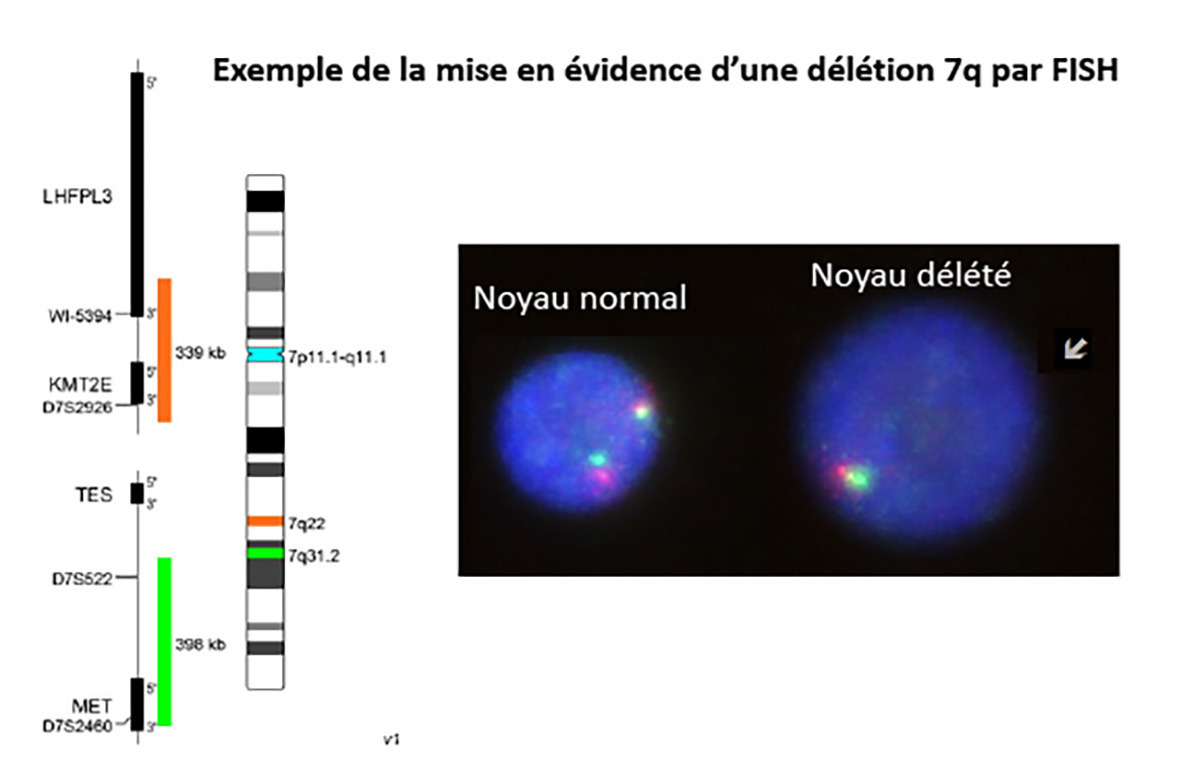
Le caryotype est réalisé après mise en culture d’une partie du prélèvement obtenu à la ponction osseuse. Il est éventuellement complété par des techniques de fluorescence in situ après hybridation par sonde fluorescente (FISH). Dans 50 % des cas, le caryotype est anormal et objective :
– des anomalies de nombre : délétions (partielles ou totales : monosomies) ou gain (partiel ou total : trisomies) ;
– des translocations (rarement équilibrées).
Les anomalies les plus fréquentes sont celles du chromosome 7 : monosomie ou délétion partielle du bras long [del(7q)], du chromosome 5 [del(5q)] et du 8 (essentiellement trisomie 8).
Le caryotype est dit « complexe » quand il dévoile au moins 3 anomalies et « monosomal » quand il met en évidence au moins 2 monosomies. Les
Biologie moléculaire
Elle est réalisée dans les centres spécialisés et repose sur les techniques de séquençage nouvelle génération (next generation sequencing, NGS) à l’aide de panels recherchant des mutations somatiques de gènes connus pour leur implication dans les SMD.
Les mutations les plus souvent observées sont impliquées dans la régulation de l’épissage (SF3B1, SRSF2), la méthylation (TET2, DNMT3A), la modulation de la chromatine (ASXL1) ou sont des facteurs de transcription (RUNX1). Environ 5 à 10 % des SMD ont une mutation de TP53, un gène suppresseur de tumeur.
Certaines de ces mutations sont pronostiques, d’autres constituent de potentielles cibles thérapeutiques, en cours d’évaluation.
Quels diagnostics différentiels ?
Aucun des résultats d’examens précédemment décrits n’est pathognomonique d’un SMD. En effet, certains virus ou toxiques sont à l’origine de dysplasie médullaire, tandis que l’âge peut favoriser une hématopoïèse clonale (des mutations somatiques dans la moelle) sans réelle myélodysplasie.
Les formes particulières de SMD sont potentiellement plus difficiles à diagnostiquer (
Par ailleurs, il existe des maladies hématologiques « frontières » avec ce syndrome, comme l’aplasie médullaire idiopathique ou les SMP, qu’il faut savoir différencier pour mieux traiter les patients. Certains examens biologiques complémentaires sont à même d’éliminer ces diagnostics. On peut ainsi citer parmi les diagnostics différentiels :
– l’aplasie médullaire, une maladie non clonale de la cellule souche hématopoïétique, caratérisée par un défaut de production de cellules sanguines. Le diagnostic est porté à l’aide d’une biopsie ostéomédullaire (BOM) ;
– le syndrome myéloprolifératif (SMP), surtout de type myélofibrose (anciennement splénomégalie myéloïde). Le diagnostic de myélofibrose est évoqué par la présence de mutations associées aux SMP (JAK2, CALR et MPL) et recherchées dans le sang ou la moelle osseuse. La fibrose s’évalue par la réalisation d’une BOM ;
– la prise de certains médicaments (antiviraux, anti- tuberculeux, immunosuppresseurs) ;
– la carence en vitamine B12 ou folates, recherchée par un dosage sanguin ;
– des infections virales (VIH, parvovirus B19) à investiguer si les signes ou l’histoire clinique sont évocateurs.
Pronostic et complications
Classification OMS
Revue en 2016, elle différencie essentiellement les SMD avec excès de blastes (MDS-EB), ceux avec excès de sidéroblastes en couronne (MDS-RS), avec del(5q), avec atteinte d’une seule (MDS-SLD) ou plusieurs (MDS-MLD) lignées, et les SMD inclassables (MDS-UD).3
Cette classification est cytologique, obtenue à partir du résultat du myélogramme. Elle est descriptive, ne tient pas compte des autres paramètres pronostiques et ne permet pas à elle seule d’orienter la décision thérapeutique.
Évaluation du risque de transformation
Le pronostic des patients est défini à l’aide du score IPSS-R (International Prognostic Scoring System-Revised 2012) [
En pratique, on distingue :
– les SMD de « faible risque » (avec un IPSS-R de risque très faible ou faible), avec une évolution vers une LAM dans 15 % des cas ;
– les SMD de « haut risque » (IPSS-R élevé ou très élevé ± les intermédiaires) ; LAM dans 30 % des cas.
D’autres facteurs pronostiques sont pris en compte, comme l’âge, l’existence de comorbidités, la dépendance transfusionelle chez les faibles risques ou la surcharge martiale.
Aggravation des cytopénies
Les complications du SMD sont en rapport avec une aggravation des cytopénies et/ou une transformation en LAM. Toutefois, compte tenu de l’âge médian de survenue des SMD, il n’est pas rare que les patients décèdent d’autres maladies, surtout dans les formes de faible risque.
L’anémie en elle-même ne présente que rarement une cause de complications mettant en jeu le pronostic vital, puisque les patients sont transfusés. Cependant, elle impacte leur qualité de vie du fait de la dépendance aux transfusions les obligeant à venir régulièrement à l’hôpital (il n’existe pas en France de possibilité de transfusion à domicile). Par ailleurs, l’anémie chronique est souvent accompagnée d’une asthénie gênante dans la vie quotidienne.
Un autre aspect indirect de l’anémie est la surcharge martiale (en fer) induite par les transfusions itératives. Elle peut être à l’origine de complications cardiaques, hépatiques et cérébrales. Elle est évaluée par le taux de ferritine et par des IRM des différents organes. Elle nécessite parfois un traitement.
La thrombopénie peut être à l’origine d’hémorragies, et fait partie des causes de décès (rares cependant et souvent en rapport avec une hémorragie cérébrale) chez les malades. Son impact sur la qualité de vie est similaire à l’anémie du fait d’une dépendance aux transfusions plaquettaires. Les symptômes hémorragiques sont rarement spontanés si le taux de plaquettes est supérieur à 20 000/mm³. Néanmoins, l’adjonction d’un traitement antiagrégant ou anticoagulant est fréquente dans cette tranche d’âge, conduisant à un risque hémorragique pour un seuil inférieur à 50 000/mm³. Le développement d’une allo-immunisation plaquettaire à la suite des transfusions itératives n’est pas rare, nécessitant l’utilisation de produits sanguins particuliers et compliquant la prise en charge.
La neutropénie est à l’origine d’infections, cause la plus fréquente de décès pour les SMD de haut risque. Elles sont essentiellement bactériennes et affectent préférentiellement les poumons, la sphère ORL ou digestive. Les infections fongiques sont plus rares, essentiellement observées à l’occasion d’une chimiothérapie ou d’une allogreffe de moelle osseuse, mais sont très sévères et difficiles à traiter du fait de la neutropénie chronique.
Lors de la transformation en LAM, le pronostic devient défavorable compte tenu de l’exposition fréquente à de longues périodes de neutropénie, et d’un faible taux de réponse aux thérapeutiques classiques en rapport avec les caractéristiques cytogénétiques et moléculaires de ces LAM post-SMD.
Traitement : 2 situations distinctes
La prise en charge doit se faire en milieu spécialisé.
Elle est guidée par le score IPSS-R.
Le seul traitement curatif des SMD aujourd’hui est l’allogreffe de cellules souches hématopoïètiques, procédure lourde réservée aux patients les plus jeunes et en l’absence de comorbidité.
SMD de faible risque
L’abstention thérapeutique avec une surveillance en milieu spécialisé est recommandée lorsque les cytopénies sont bien tolérées et modérées. Lorsqu’elles deviennent sévères et/ou symptomatiques, elles sont traitées :
– les transfusions de produits sanguins labiles sont délivrées en hôpital de jour. Deux culots globulaires sont classiquement dispensés pour maintenir un taux d’hémoglobine minimal autour de 8 g/dL. Ce seuil varie en fonction de la tolérance individuelle de chaque patient à l’anémie. Le seuil plaquettaire est généralement de 20 000/mm³ (cf. supra) pour les transfusions de culots plaquettaires, mais peut être plus élevé en fonction des symptômes hémorragiques observés ou si des traitements agissant sur la coagulation (anticoagulants ou antiagrégants plaquettaires) sont coprescrits ;
– les agents stimulants l’érythropoïèse.8 L’érythropoïètine (EPO) est indiquée en première intention en traitement de l’anémie symptomatique ou nécessitant des transfusions. Plusieurs EPO recombinantes ou des dérivés sont disponibles (EPO-alfa [Binocrit, Eprex]; EPO-bêta [Neorecormon], darbépoétine [Aranesp], EPO-zêta [Retacrit]) mais la seule ayant l’AMM est Aranesp. Elles sont le plus souvent prescrites en injection sous-cutanée hebdomadaire, avec une surveillance de l’hémogramme, pour un objectif de concentration en hémoglobine entre 11 et 12 g/dL. Le taux de réponse observé est entre 30 et 50 %, et la durée médiane de réponse est de 2 ans ;
– Revlimid (lénalidomide), traitement immuno- modulateur, est indiqué dans le traitement de l’anémie chez les patients avec un syndrome 5q- ;
– en cas d’échec de ces traitements pour l’anémie, il n’existe pas de médicament ayant l’AMM aujourd’hui. On peut cependant citer le luspatercept, agissant via la voie du TGF-bêta et ciblant l’érythropoïèse tardive (l’EPO ciblant l’érythropoïèse précoce). Il est en cours d’évaluation et donne des résultats encourageants dans les essais cliniques ;
– la thrombopénie est essentiellement prise en charge par des transfusions, mais des analogues de la thrombopoïétine sont actuellement en cours de développement, et semblent avoir une efficacité chez les patients thrombopéniques de faible risque.
SMD de haut risque
Quand le patient est en âge de pouvoir recevoir une allogreffe de cellules souches hématopoïétiques (moins de 70 ans), elle doit être envisagée. Les autres approches ont pour objectif de contrôler la maladie en limitant les cytopénies, mais aussi de retarder la transformation en LAM :
– les agents hypométhylants :8 azacitidine (Vidaza). C’est le principe actif de référence des SMD de haut risque. Le mécanisme d’action repose sur la réduction du taux de méthylation de l’ADN, souvent élevé dans cette pathologie et dans les LAM. Cette hyperméthylation de l’ADN intervient dans la physiopathologie des SMD, aboutissant à l’inhibition de l’expression de certains gènes suppresseurs de tumeur. Le taux de réponse hématologique est de 50 à 60 %, avec une durée de réponse de 13 à 15 mois. Ce traitement est administré en sous-cutané, habituellement pendant 7 jours consécutifs tous les 28 jours. Les effets secondaires sont essentiellement hématologiques, avec une majoration des cytopénies lors des premiers cycles (impliquant de facto une augmentation du risque infectieux), avant l’obtention de la réponse hématologique survenant le plus souvent entre le 3e et le 4e cycle ;
– la chimiothérapie intensive est moins utilisée depuis l’avènement des hypométhylants. Elle est plutôt réalisée dans les formes avec un excès de blastes proches de la LAM, à caryotype normal, et en vue d’une allogreffe de cellules souches hématopoïétiques ;
– l’allogreffe de cellules souches hématopoïétiques9 est curative, limitée en général aux patients de moins de 70 ans en l’absence de comorbidités et avec un donneur. Le plus souvent, les conditionnements sont atténués, ce qui signifie que le receveur, en préparation de la greffe, reçoit un traitement non myélo-ablatif, moins toxique qu’un conditionnement standard. La place des thérapeutiques en prégreffe ou en entretien post-greffe demeure controversée ;
– d’autres molécules sont en cours d’évaluation dans les SMD, souvent en association avec Vidaza. Le médicament actuellement le plus avancé dans le développement thérapeutique et ayant le plus d’efficacité est le vénétoclax (Venclyxto). Il agit en bloquant la protéine BCL-2. Il pourrait apporter un bénéfice thérapeutique en association avec Vidaza dès la première ligne de traitement ;
– dans certaines formes frontières avec l’aplasie médullaire, un traitement immunologique par ciclo–sporine et sérum antilymphocytaire (cheval) peut être proposé.
Remerciements à : Stéphanie Mathis et Giulia Tueur (laboratoire d’hématologie de l’hôpital Saint-Louis) pour les illustrations de cytologie et de cytogénétique.
1. Quand penser à un SMD ?
Devant un patient âgé de plus de 70 ans.
Ayant été traité pour un cancer par radio- ou chimiothérapie.
Et/ou exposé aux benzènes ou solvants dans sa vie professionnelle.
Essouflé, fatigué, ayant des saignements ou une infection bactérienne.
Avec un hémogramme anormal : cytopénie(s), monocytes circulants > 10 %.
2. Formes particulières de SMD
Le syndrome 5q- atteint surtout les femmes à partir de 60 ans.5 L’anémie est souvent importante, macrocytaire, non régénérative, et les patients ont également une hyperplaquettose (jusqu’à 1 000 000/mm³). Le myélogramme montre un aspect particulier, avec des mégacaryocytes géants et unilobés. La cytogénétique révèle une délétion du bras long du chromosome 5 isolée mettant toujours en cause la région 5q31.1 (région commune délétée), contenant le gène RPS14, impliqué dans la voie P53. Le pronostic est plutôt favorable comparé aux autres SMD, et il existe un traitement spécifique : le lénalidomide (Revlimid) avec plus de 80 % de réponse.
La leucémie myélomonocytaire chronique (LMMC) est un syndrome frontière SMD/SMP. Longtemps classé parmi les SMD, la LMMC associe une prolifération monocytaire (> 10 % des leucocytes), l’absence de chromosome Philadelphie, une dysplasie médullaire d’au moins 1 lignée et un éventuel excès de blastes médullaires (< 20 %) ou sanguins (< 10 %). Elle est souvent associée à une splénomégalie. Un traitement par hydroxyurée ou par des agents hypométhylants est envisageable.
1. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364:2496-506
2. Neukirchen J, Schoonen WM, Strupp C, et al. Incidence and prevalence of myelodysplastic syndromes: data from the Düsseldorf MDS-registry. Leuk Res 2011;35:1591-6.
3. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009;114:937-51.
4. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012;120:2454-65.
5. Boultwood J, Fidler C, Strickson AJ, et al. Narrowing and genomic annotation of the commonly deleted region of the 5q- syndrome. Blood 2002;99:4638-41.
6. Jädersten M, Malcovati L, Dybedal I, et al. Erythropoietin and granulocyte-colony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol 2008;26:3607-13.
7. List A, Dewald G, Bennett J, et al, for the Myelodysplastic Syndrome-003 Study Investigators. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med 2006;355:1456-65.
8. FenauxP,MuftiGJ,Hellstrom-LindbergE,etal,forthe International Vidaza High-Risk MDS Survival Study Group. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009;10:223-32.
9. Sierra J, Pérez WS, Rozman C, et al. Bone marrow transplantation from HLA-identical siblings as treatment for myelodysplasia. Blood 2002;100:1997-2004.
Dans cet article
 Encadrés
Encadrés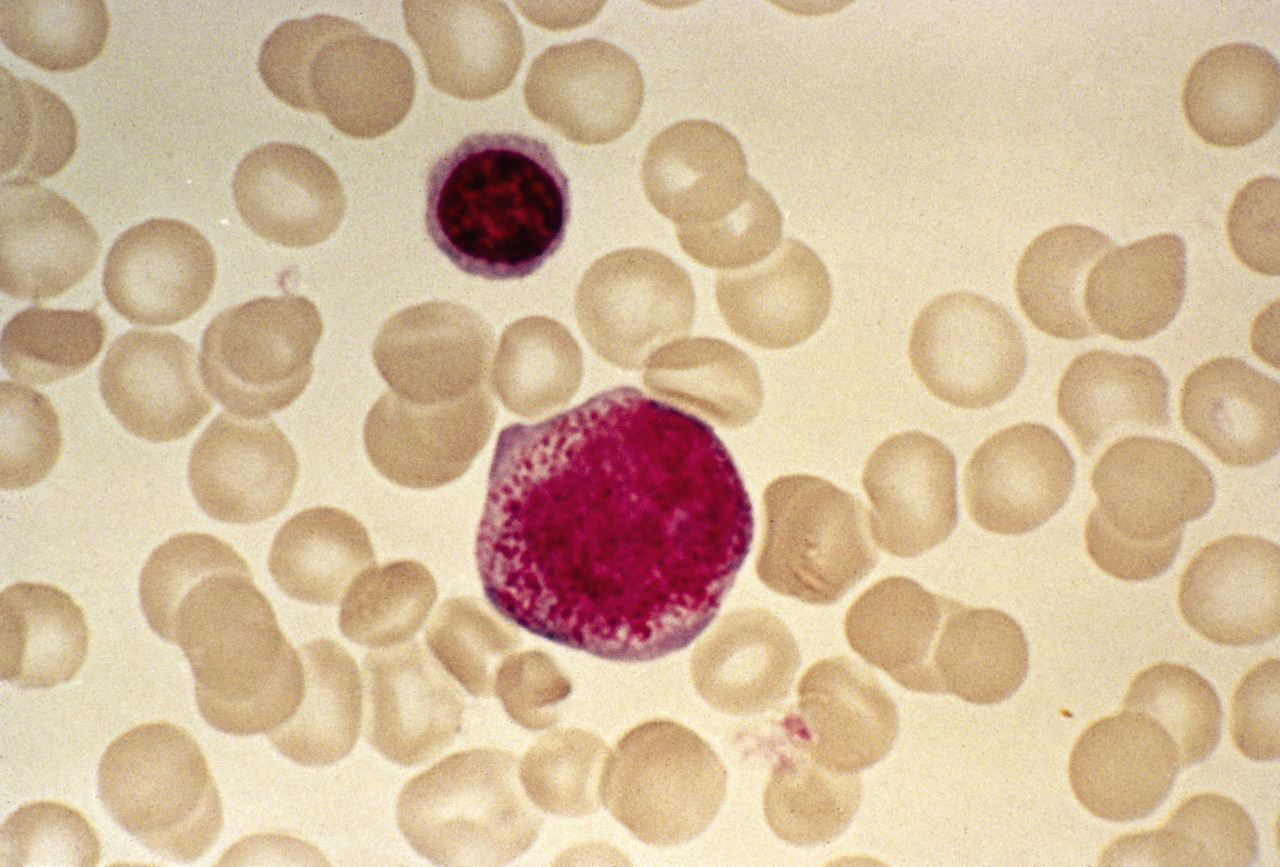






Le SMD est une maladie clonale, hétérogène, touchant le sujet âgé.
Il est le plus souvent idiopathique (80 % des cas), parfois secondaire à une exposition à des chimio-/radiothérapies ou à une prédisposition génétique.
Les signes cliniques les plus fréquents sont en rapport avec une anémie, parfois découverte de manière fortuite sur un hémogramme systématique.
C’est le myélogramme qui fait le diagnostic, en objectivant des signes de dysplasie dans la moelle en cytologie, et parfois un excès de cellules immatures : les blastes.
La classification pronostique est fondée sur les paramètres de l’hémogramme, le pourcentage de blastes médullaires et la cytogénétique (caryotype anormal dans 50 % des cas).
Il existe principalement 2 catégories : les SMD de faible risque qui posent le problème des cytopénies (essentiellement l’anémie) et les SMD de haut risque, qui doivent être traités plus intensivement pour limiter le risque de transformation en LAM.
La prise en charge de l’anémie repose sur la prescription d’EPO et les transfusions de culots globulaires.
Le traitement des SMD de haut risque s’appuie sur l’azacitidine (Vidaza), un hypométhylant.
L’allogreffe de cellules souches hématopoïètiques est le seul traitement curatif des SMD, mais ne peut être proposée qu’à de rares patients.