Devant une hyperéosinophilie sanguine, la démarche du clinicien est double : évaluer son retentissement sur les organes, et déterminer sa cause ; mais 50 % des syndromes hyperéosinophiliques restent d’origine indéterminée.
Les syndromes hyperéosinophiliques (SHE) – définis par l’association d’une hyper- éosinophilie sanguine supérieure ou égale à 1,5 G/L d’évolution chronique (> 1 mois) associée à des dommages tissulaires en rapport avec l’infiltration éosinophilique (tableau 1 ) – sont une entité hétérogène aux mécanismes physiopathologiques sous-jacents variés. Le terme de « syndrome hyperéosinophilique » a été proposé pour la première fois en 1968, et un premier jeu de critères de classification a été suggéré en 1975.1 À mesure de l’amélioration des connaissances fondamentales relatives aux processus physiopathologiques sous-jacents de l’éosinophilie (notamment la découverte chez certains patients d’anomalies hématopoïétiques clonales ou la présence de lymphocytes T clonaux de phénotype anormal), des actualisations successives de ces critères ont été proposées.
Classification et principaux sous-types
Actuellement, la principale classification des SHE utilisée pour le soin et la recherche est celle proposée en 2011 par le Groupe de travail coopératif international sur les maladies à éosinophiles (ICOG-EO, comprenant un panel multidisciplinaire d’experts immunologistes, allergologues, hématologues et anatomopathologistes).2 Ainsi, on distingue : les SHE clonaux (SHEn), dont la leucémie chronique à éosinophiles liée à la délétion FIP1L1-PDGFRA (LCE F/P+) et les éosinophilies associées aux autres syndromes myéloprolifératifs et myélodysplasiques (tableau 2 ) ; les SHE réactionnels (SHEr), où les éosinophiles – polyclonaux – sont induits par la sécrétion en excès d’interleukine 5 (IL-5) qui est la cyto-kine essentielle à la maturation médullaire, la migration et l’activation des éosinophiles. Ces SHEr peuvent être d’origines diverses, notamment iatrogènes (syndrome d’hypersen- sibilité médicamenteuse retardée), infectieuses (helminthiase), ou être la conséquence d’un cancer solide (adénocarcinome bronchique ou prostatique notamment) ou hématologique (lymphome de Hodgkin, lymphomes T) [tableau 3]. Parmi les SHEr, on distingue plus particulièrement les SHE lymphoïdes (SHEl), où la production d’IL-5 est liée à la présence d’une lymphoprolifération T de bas grade de phénotype aberrant (généralement CD3-CD4+).3 Malgré un bilan causal exhaustif large, on estime qu’environ 50 % des SHE restent d’origine indéterminée : on parle alors de SHE idiopathique (SHEi).4
De même, il existe un certain nombre de pathologies « spécifiques » d’organes liées aux éosinophiles (myocardite à éosinophiles, gastro-entérite à éosinophiles, pneumopathie aiguë et/ou chronique à éosinophiles…) qui peuvent survenir isolément ou s’intégrer dans le cadre d’un SHE systémique. Enfin, la frontière nosologique est parfois ténue entre les SHE et certains syndromes tels que la granulomatose éosinophilique avec polyangéite (anciennement syndrome de Churg-Strauss).
De même, il existe un certain nombre de pathologies « spécifiques » d’organes liées aux éosinophiles (myocardite à éosinophiles, gastro-entérite à éosinophiles, pneumopathie aiguë et/ou chronique à éosinophiles…) qui peuvent survenir isolément ou s’intégrer dans le cadre d’un SHE systémique. Enfin, la frontière nosologique est parfois ténue entre les SHE et certains syndromes tels que la granulomatose éosinophilique avec polyangéite (anciennement syndrome de Churg-Strauss).
Manifestations cliniques
Que ce soit via la libération de protéines cytotoxiques (telles les protéines cationiques des éosinophiles comme la protéine basique majeure [MBP], la peroxydase de l’éosinophile [EPO]…), de dérivés réactifs de l’oxygène, de cytokines pro-inflammatoire (IL-1, IL-6, Tumor Necrosis Factor alpha [TNF-α]), de facteurs pro-fibrosants (tels l’IL-13 et le Transforming Growth Factor beta [TGF-β]) ou de facteurs pro-coagulants, les polynucléaires éosinophiles exercent des effets pléïomorphes délétères sur leurs tissus cibles. De plus, il est important de signaler que la sévérité des manifestations n’est pas proportionnelle à l’importance de l’hypéréosinophilie sanguine, mais plus en lien avec l’importance de l’infiltrat tissulaire et le degré d’activation des polynucléaires éosinophiles au sein de ces tissus. Du fait de la diversité des sous-types de SHE et des mécanismes sous-jacents à l’hyperéosinophilie, il est difficile de brosser un tableau homogène de ce syndrome protéiforme.
Dans la littérature, les séries rapportées sont généralement de faibles effectifs et influencées par la nature du recrutement (hématologique, dermatologique…). À ce jour, la plus large série publiée est issue d’un processus de coopération internationale transdisciplinaire, et porte sur 188 patients aux profils variés.4 Le sex-ratio est proche de 1, l’âge de début se situe habituellement entre 20 et 50 ans et les signes généraux sont d’ordinaire peu intenses (en accord avec l’absence de syndrome inflammatoire marqué). Par ordre de fréquence, les organes les plus fréquemment atteints sont la peau (69 %), les poumons (44 %) et le tube digestif (38 %).4
Les manifestations dermatologiques sont polymorphes (prurit, éruptions maculo-papuleuses ou eczématiformes) bien que certaines (hémorragies sous-unguéales en flammèches, ulcérations muqueuses buccales et/ou génitales, angiœdèmes) soient très évocatrices de SHE.
Les manifestations respiratoires associent atteintes bronchiques (asthme hyperéosinophilique, voire bronchiolite oblitérante hyperéosinophilique) et parenchymateuses (pneumopathies aiguës ou chroniques à éosinophiles), avec également des formes de chevauchement possibles avec la granulomatose éosinophilique avec polyangéite (en l’absence d’anticorps anti-cytoplasme des neutrophiles [ANCA]). Dans ce contexte, signalons l’apport potentiel du dosage de la protéine C-réactive (CRP) qui pourrait permettre de différencier les patients ayant un SHE (où les taux de CRP sont généralement bas) de ceux souffrant d’une vascularite systémique (où les signes inflammatoires sont généralement plus marqués).5
Les manifestations oto-rhino-laryngées, allant souvent de pair avec l’atteinte respiratoire, et pouvant constituer la triade de Widal (association d’un asthme, d’une intolérance à l’aspirine ou aux anti-inflammatoires non stéroïdiens et d’une polypose nasale), peuvent être responsables d’un inconfort majeur.
L’ensemble du tube digestif peut être touché : si l’œsophagite à éosinophiles – d’origine allergique vraisemblable quand elle est isolée – est généralement responsable d’une dysphagie invalidante et/ou d’impactions alimentaires chez l’enfant ou l’adolescent, les manifestations gastro-entéritiques sont plus variées, associant douleurs abdominales, diarrhées et/ou ascite (notamment en cas d’atteinte séreuse).
L’atteinte cardiaque, initialement considérée comme fréquente, semble n’affecter en réalité que 20 % des patients.4 Celle-ci évolue en plusieurs phases : initialement, il s’agit d’une myocardite à éosinophiles pouvant menacer le pronostic vital (insuffisance cardiaque aiguë, troubles de conduction) ; dans un 2e temps, développement de thrombus (potentiellement emboligènes) sur l’endocarde avec finalement un risque d’évolution vers une cardiopathie restrictive (par fibrose endomyocardique) ou dilatée séquellaire.
D’autres manifestations sont plus rares, voire exceptionnelles : atteintes neurologiques centrales (focales ou non) ou périphériques (polyneuropathies à prédominance sensitive), urologiques (cystites à éosinophiles), biliaires (cholangites à éosinophiles), manifestations thromboemboliques veineuses et/ou artérielles, ou vasculopathies distales « pseudo-Buerger ».
Plus généralement, le groupe de travail coopératif international ICOG-EO a établi en 2011 que toute manifestation dermatologique (y compris muqueuse), neurologique (centrale ou périphérique), thrombotique (veineuse ou artérielle), fibrosante (quel que soit le site), ou que toute « atteinte d’organe étant la conséquence présumée de la toxicité des éosinophiles » survenant chez un patient ayant une hyperéosinophilie sanguine supérieure à 1,5 G/L chronique (> 1 mois) permettait de classer ce patient comme ayant un SHE.2 Cette définition, bien que très souple et élargissant considérablement le spectre des SHE, a néanmoins le mérite de sensibiliser les cliniciens (quelle que soit leur spécialité) sur le fait qu’ils sont parfois confrontés – chez des patients ayant des manifestations d’organe inexpliquées et une hyperéosinophilie sanguine – à un authentique SHE. Il faut alors s’astreindre au bilan étiologique du SHE, et se poser sans tarder la question du traitement.
Dans la littérature, les séries rapportées sont généralement de faibles effectifs et influencées par la nature du recrutement (hématologique, dermatologique…). À ce jour, la plus large série publiée est issue d’un processus de coopération internationale transdisciplinaire, et porte sur 188 patients aux profils variés.4 Le sex-ratio est proche de 1, l’âge de début se situe habituellement entre 20 et 50 ans et les signes généraux sont d’ordinaire peu intenses (en accord avec l’absence de syndrome inflammatoire marqué). Par ordre de fréquence, les organes les plus fréquemment atteints sont la peau (69 %), les poumons (44 %) et le tube digestif (38 %).4
Les manifestations dermatologiques sont polymorphes (prurit, éruptions maculo-papuleuses ou eczématiformes) bien que certaines (hémorragies sous-unguéales en flammèches, ulcérations muqueuses buccales et/ou génitales, angiœdèmes) soient très évocatrices de SHE.
Les manifestations respiratoires associent atteintes bronchiques (asthme hyperéosinophilique, voire bronchiolite oblitérante hyperéosinophilique) et parenchymateuses (pneumopathies aiguës ou chroniques à éosinophiles), avec également des formes de chevauchement possibles avec la granulomatose éosinophilique avec polyangéite (en l’absence d’anticorps anti-cytoplasme des neutrophiles [ANCA]). Dans ce contexte, signalons l’apport potentiel du dosage de la protéine C-réactive (CRP) qui pourrait permettre de différencier les patients ayant un SHE (où les taux de CRP sont généralement bas) de ceux souffrant d’une vascularite systémique (où les signes inflammatoires sont généralement plus marqués).5
Les manifestations oto-rhino-laryngées, allant souvent de pair avec l’atteinte respiratoire, et pouvant constituer la triade de Widal (association d’un asthme, d’une intolérance à l’aspirine ou aux anti-inflammatoires non stéroïdiens et d’une polypose nasale), peuvent être responsables d’un inconfort majeur.
L’ensemble du tube digestif peut être touché : si l’œsophagite à éosinophiles – d’origine allergique vraisemblable quand elle est isolée – est généralement responsable d’une dysphagie invalidante et/ou d’impactions alimentaires chez l’enfant ou l’adolescent, les manifestations gastro-entéritiques sont plus variées, associant douleurs abdominales, diarrhées et/ou ascite (notamment en cas d’atteinte séreuse).
L’atteinte cardiaque, initialement considérée comme fréquente, semble n’affecter en réalité que 20 % des patients.4 Celle-ci évolue en plusieurs phases : initialement, il s’agit d’une myocardite à éosinophiles pouvant menacer le pronostic vital (insuffisance cardiaque aiguë, troubles de conduction) ; dans un 2e temps, développement de thrombus (potentiellement emboligènes) sur l’endocarde avec finalement un risque d’évolution vers une cardiopathie restrictive (par fibrose endomyocardique) ou dilatée séquellaire.
D’autres manifestations sont plus rares, voire exceptionnelles : atteintes neurologiques centrales (focales ou non) ou périphériques (polyneuropathies à prédominance sensitive), urologiques (cystites à éosinophiles), biliaires (cholangites à éosinophiles), manifestations thromboemboliques veineuses et/ou artérielles, ou vasculopathies distales « pseudo-Buerger ».
Plus généralement, le groupe de travail coopératif international ICOG-EO a établi en 2011 que toute manifestation dermatologique (y compris muqueuse), neurologique (centrale ou périphérique), thrombotique (veineuse ou artérielle), fibrosante (quel que soit le site), ou que toute « atteinte d’organe étant la conséquence présumée de la toxicité des éosinophiles » survenant chez un patient ayant une hyperéosinophilie sanguine supérieure à 1,5 G/L chronique (> 1 mois) permettait de classer ce patient comme ayant un SHE.2 Cette définition, bien que très souple et élargissant considérablement le spectre des SHE, a néanmoins le mérite de sensibiliser les cliniciens (quelle que soit leur spécialité) sur le fait qu’ils sont parfois confrontés – chez des patients ayant des manifestations d’organe inexpliquées et une hyperéosinophilie sanguine – à un authentique SHE. Il faut alors s’astreindre au bilan étiologique du SHE, et se poser sans tarder la question du traitement.
Quelle exploration ? Quel bilan étiologique ?
Les examens complémentaires proposés à un patient ayant une hyperéosinophilie sanguine ont deux finalités : évaluer son retentissement systémique, et en chercher la cause. Ces deux démarches doivent être menées conjointement et sont orientées par les données de l’interrogatoire, de l’examen clinique, et des principaux résultats biologiques de routine.
L’hyperéosinophilie est modérée
Devant une hyperéosinophilie modérée (entre 0,5 et 1,5 G/L), on évoque en premier l’atopie, les parasitoses sans cycle tissulaire (oxyure, tænia, gale), l’insuffisance surrénale lente ou l’infection par le virus de l’immunodéficience humaine (VIH).
L’hyperéosinophilie est supérieure à 1,5 G/L
Devant une hyperéosinophilie > 1,5 G/L, le patient doit bénéficier :
– d’un bilan minimal, à la recherche d’une atteinte viscérale liée à l’hyperéosinophilie et incluant ionogramme sanguin, bilan hépatique complet, dosages de troponine, Brain Natriuretic Peptide (BNP), taux de prothrombine, CRP, créatine phospho-kinase, bandelette urinaire et électrocardiogramme (à réaliser en semi-urgence, selon le contexte clinique et évolutif aigu ou chronique) ;6
– d’un bilan étiologique minimal de débrouillage, avec notamment des sérologies infectieuses (virus de l’immunodéficience humaine [VIH] et toxocarose systématiques) ;
– d’autres sérologies spécifiques d’helminthiases ou de l’human T-cell lymphotropic virus de type I (HTLV1), en fonction du contexte et des anté-cédents de voyages en zone d’endémie, d’un examen parasitologique des selles (recherche d’ascaris, éventuellement à compléter par la technique de Baermann en cas d’antécédent de séjour en zone d’endémie d’anguillulose et/ou de scotch-test en cas de suspicion d’oxyurose), des dosages de tryptase (mastocytose), de vitamine B12 (l’élévation de la B12 et de la tryptase oriente vers un SHEn), d’immunoglobulines de type E (IgE) totales (en cas d’élévation, oriente vers une origine réactionnelle Th2-médiée à l’éosinophilie) et des lactates deshydrogénases (LDH) (en cas d’élévation, oriente vers une hémopathie lymphoïde), d’une électrophorèse des protéines plasmatiques (en cas d’hypergammaglobulinémie polyclonale, oriente vers une maladie à IgG4, un lymphome angio-immunoblastique ou une infection parasitaire chronique ; en cas d’hypogammaglobulinémie, oriente vers un déficit immunitaire ou une hémopathie lymphoïde) ainsi que d’un phénotypage lymphocytaire (avec mention de recherche spécifique de clone T au phénotype anormal de type CD3-CD4+ ; ou CD3+CD4+CD7- ; ou CD3+CD4-CD8-TCRab).
– d’un bilan minimal, à la recherche d’une atteinte viscérale liée à l’hyperéosinophilie et incluant ionogramme sanguin, bilan hépatique complet, dosages de troponine, Brain Natriuretic Peptide (BNP), taux de prothrombine, CRP, créatine phospho-kinase, bandelette urinaire et électrocardiogramme (à réaliser en semi-urgence, selon le contexte clinique et évolutif aigu ou chronique) ;6
– d’un bilan étiologique minimal de débrouillage, avec notamment des sérologies infectieuses (virus de l’immunodéficience humaine [VIH] et toxocarose systématiques) ;
– d’autres sérologies spécifiques d’helminthiases ou de l’human T-cell lymphotropic virus de type I (HTLV1), en fonction du contexte et des anté-cédents de voyages en zone d’endémie, d’un examen parasitologique des selles (recherche d’ascaris, éventuellement à compléter par la technique de Baermann en cas d’antécédent de séjour en zone d’endémie d’anguillulose et/ou de scotch-test en cas de suspicion d’oxyurose), des dosages de tryptase (mastocytose), de vitamine B12 (l’élévation de la B12 et de la tryptase oriente vers un SHEn), d’immunoglobulines de type E (IgE) totales (en cas d’élévation, oriente vers une origine réactionnelle Th2-médiée à l’éosinophilie) et des lactates deshydrogénases (LDH) (en cas d’élévation, oriente vers une hémopathie lymphoïde), d’une électrophorèse des protéines plasmatiques (en cas d’hypergammaglobulinémie polyclonale, oriente vers une maladie à IgG4, un lymphome angio-immunoblastique ou une infection parasitaire chronique ; en cas d’hypogammaglobulinémie, oriente vers un déficit immunitaire ou une hémopathie lymphoïde) ainsi que d’un phénotypage lymphocytaire (avec mention de recherche spécifique de clone T au phénotype anormal de type CD3-CD4+ ; ou CD3+CD4+CD7- ; ou CD3+CD4-CD8-TCRab).
D’autres examens sont fonction du contexte
Les autres examens (y compris morphologiques, endoscopiques et bio-psiques) sont orientés par le contexte : ainsi, en cas d’atteinte bronchique, on complète ce bilan généralement par la recherche d’ANCA et une sérologie aspergillaire avec dosages des IgE totales et spécifiques anti-aspergillaires (afin d’étayer les diagnostics éventuels de granulomatose éosinophilique avec polyangéite ou d’aspergillose broncho-pulmonaire allergique). En cas d’atteinte cutanée prépondérante, on peut, en fonction de la nature des lésions dermatologiques, rechercher des cellules de Sézary (au frottis sanguin) ou la présence d’anticorps anti-membrane basale (dans l’hypothèse d’une pemphigoïde bulleuse).
Le patient a un phénotype orientant vers un SHEn
Enfin, quand le patient a un phénotype orientant vers un SHEn (hépato-splénomégalie, autre anomalie de l’hémogramme, augmentation de la vitamine B12 ou de la tryptase, corticorésistance), on cherche la présence de la délétion interstitielle FIP1L1-PDGFRA dans le sang (qui est donc un examen complémentaire dont la prescription doit rester ciblée) et, en cas de négativité, on peut réaliser un myélogramme avec caryotype (à la recherche de réarrangements chromosomiques impliquant PDGFRB, FGFR1, JAK2 ou FLT3), voire la recherche d’anomalies moléculaires du « panel myéloïde » grâce au séquençage haut débit. Dans les autres situations, les formes idiopathiques étant de loin les plus fréquentes, il n’y a pas de place au bilan initial pour les explorations médullaires.
Qui traiter ?
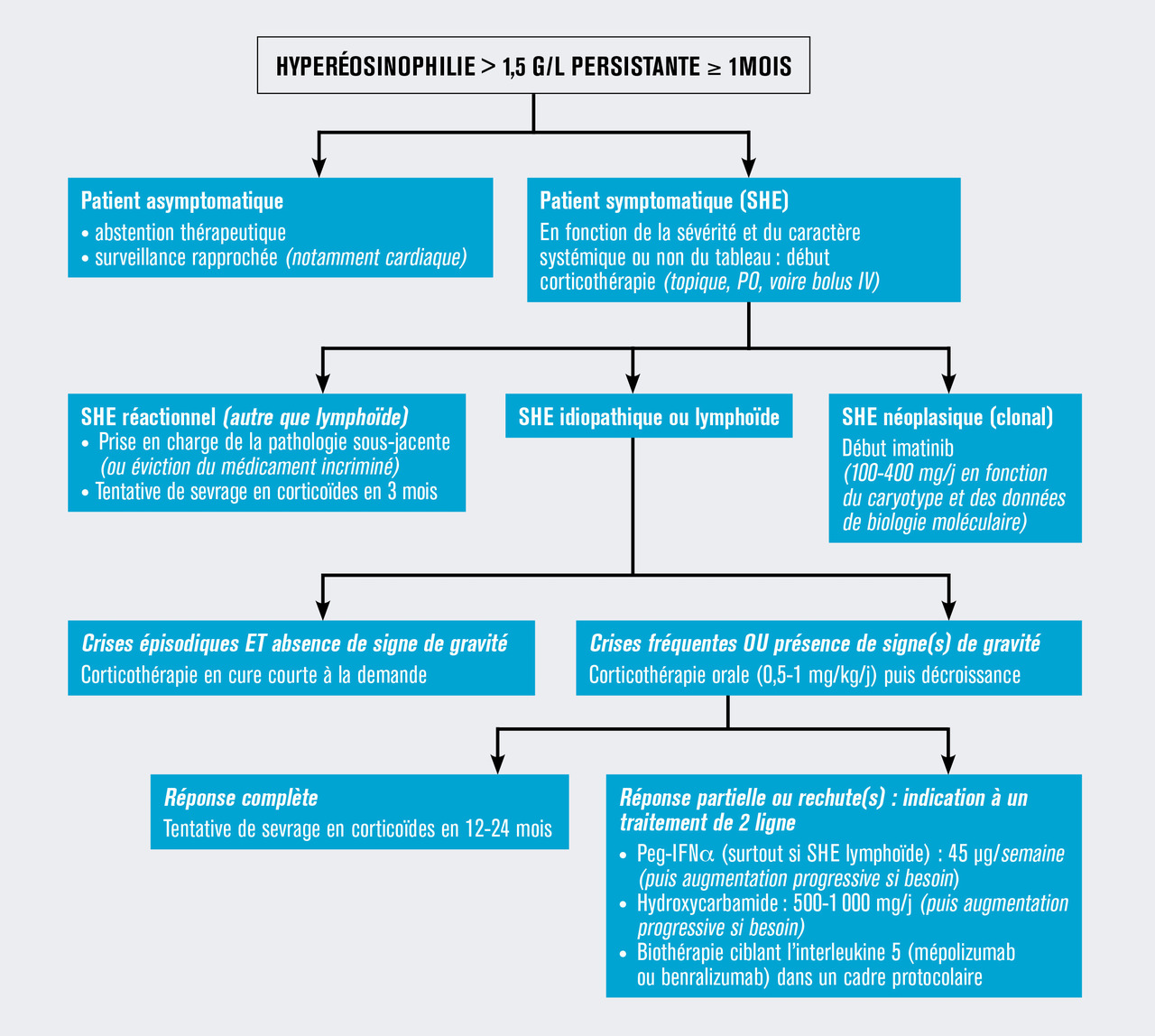
Il semble raisonnable de traiter tous les patients, symptomatiques ou non, ayant un SHEn en raison de la plus grande fréquence des atteintes viscérales (notamment cardiaque) et du risque de transformation en leucémie aiguë myéloblastique à long terme (v. figure ).7 Pour les patients atteints de SHEi ou de SHEl, les indications et modalités de traitement sont à définir au cas par cas en fonction de la gravité de l’atteinte d’organe(s) et du profil évolutif de la maladie (poussées itératives entrecoupées de périodes de rémission versus évolution chronique).
Enfin, chez les patients ayant une hyperéosinophilie (HE) asymptomatique, sans cause retrouvée, et en l’absence d’argument pour une HEn ou pour une HEl, l’importance d’une hyperéosinophilie persistante ne doit pas être un motif d’initiation de traitement. En effet, certains patients (définis comme ayant une « hyperéosinophilie de signification indéterminée » selon la classification ICOG-EO) peuvent garder une hyperéosinophilie pendant plusieurs années sans aucun retentissement clinique.8 Une surveillance rapprochée (et notamment cardiaque) est en revanche absolument nécessaire.
Enfin, chez les patients ayant une hyperéosinophilie (HE) asymptomatique, sans cause retrouvée, et en l’absence d’argument pour une HEn ou pour une HEl, l’importance d’une hyperéosinophilie persistante ne doit pas être un motif d’initiation de traitement. En effet, certains patients (définis comme ayant une « hyperéosinophilie de signification indéterminée » selon la classification ICOG-EO) peuvent garder une hyperéosinophilie pendant plusieurs années sans aucun retentissement clinique.8 Une surveillance rapprochée (et notamment cardiaque) est en revanche absolument nécessaire.
Comment traiter ?
La meilleure compréhension des mécanismes moléculaires à l’origine des SHE a permis le développement de thérapies ciblées (notamment les inhibiteurs de tyrosine kinase et les anticorps monoclonaux ciblant l’IL-5) et a considérablement modifié leur prise en charge. Si cette dernière demeure complexe et est souvent décidée au cas par cas, un programme national de diagnostic et de soins est en cours d’élaboration, et devrait permettre d’aborder les principales situations rencontrées.
Dans un contexte d’urgence
Dans un contexte d’urgence (myo-cardite, asthme aigu grave, atteinte neurologique centrale ou périphérique…), et quelle que soit la cause du SHE (y compris parasitaire, à l’exclusion de l’anguillulose maligne), la prise en charge initiale d’une atteinte viscérale sévère liée à la toxicité des éosinophiles repose sur la corticothérapie (prednisone : 0,5-1 mg/kg/j selon la gravité). En outre, dans les SHEr, le traitement doit également s’appuyer sur le traitement de la pathologie sous-jacente ou l’éviction du médicament incriminé. En cas de bilan étiologique négatif, un traitement antiparasitaire d’épreuve est généralement proposé (à distance de la phase aiguë en cas d’atteinte viscérale grave, notamment cardiaque) dans les SHE d’origine indéterminée (SHEi). De plus, un traitement par ivermectine doit systématiquement être mis en place en prévention de l’anguillulose maligne chez les patients chez qui une corticothérapie générale est indiquée et ayant un antécédent de séjour en zone d’endémie, quelle qu’en soit l’ancienneté.
Au long cours
En cas de LCE F/P+, l’imatinib mésylate (premier inhibiteur de tyrosine kinase développé) est, à la posologie de 100 mg/j, d’une efficacité quasi constante, permettant l’obtention d’une rémission hématologique rapide (< 15 jours, bien que la réponse moléculaire puisse prendre plusieurs mois).9 Chez les patients en rémission moléculaire prolongée (> 24 mois), bien qu’il n’y ait pas d’attitude codifiée, une tentative d’arrêt de l’imatinib mésylate est possible, bien que des rechutes surviennent dans environ 50% des cas, généralement dans les 2 ans suivant l’interruption du traitement. Fort heureusement, la reprise du traitement antérieur est alors généralement à nouveau efficace.9
Dans les SHEn impliquant d’autres tyrosine kinases (tel le transcrit ETV6-PDGFRB résultant de la translocation 5-12), l’imatinib mésylate est également indiqué mais à des posologies supérieures, semblables à celles utilisées dans la leucémie myéloïde chronique (soit 400 mg/j). Enfin, chez des patients ayant un SHE corticorésistant sans anomalie cytogénétique identifiée, mais ayant un « phénotype » compatible avec celui d’un syndrome myéloprolifératif, un test thérapeutique empirique avec l’imatinib mésylate peut également être proposé ponctuellement en raison de la possible implication d’autres tyrosine kinases non identifiables par les outils de biologie moléculaire actuels.10
Dans les situations autres que les SHEn, en cas d’inefficacité ou de corticodépendance à un seuil élevé, les traitements de deuxième ligne sont l’hydroxyurée et le peginterféron alpha-2a (PEG-IFNα), qui sont parfois même utilisés en association. Les principales limites du PEG-IFNα sont sa mauvaise tolérance au long cours (syndrome pseudo-grippal post-injection, troubles neuropsychiques), mais celle-ci peut être améliorée en débutant par des posologies faibles (par exemple 45 à 90 µg hebdomadaires de PEG-IFNα). Au contraire, l’hydroxyurée est généralement bien tolérée, bien que des toxicités (notamment hématologique ou cutanée) soient possibles.
Au cours des SHEr, des SHEl et des SHEi, les biothérapies ciblant l’IL-5 sont des options très séduisantes en raison de leur physiopathologie et du caractère « IL-5-dépendant » de l’éosinophilie. Dans une étude randomisée en double aveugle contre placebo chez 85 patients atteints de SHE F/P- corticodépendant, le mépolizumab (une IgG1 kappa murine « humanisée » qui neutralise l’IL-5 circulante) à la dose de 750 mg/mois par voie intraveineuse induisait une épargne cortisonique significative et un meilleur contrôle de l’hyperéosinophilie,11 et ce avec un profil de tolérance excellent. Malgré ces résultats prometteurs et ceux rapportés dans d’autres affections voisines (telles la polypose nasosinusienne et la granulomatose éosinophilique avec polyangéite), le mépolizumab n’a pas l’autorisation de mise sur le marché pour les SHE. Dans les SHE, une deuxième étude internationale prospective randomisée évaluant ce médicament (à la dose sous-cutanée de 300 mg mensuels) contre placebo est en cours. Plus récemment, une étude de phase II portant sur 20 patients ayant un SHE non lié à FIP1L1-PDGFRA et évaluant le benralizumab (un anticorps murin bloquant la sous-unité α du récepteur à l’IL-5) a montré des résultats prometteurs.12
Enfin, de manière plus anecdotique, de nombreux autres immunosuppresseurs (ciclosporine, méthotrexate, étoposide, vincristine) ont également été essayés au cours des SHE. Chez les patients multiréfractaires, l’alemtuzumab (anticorps monoclonal ciblant le CD-52), la cladribine et l’allogreffe de moelle sont les thérapeutiques de dernière ligne à envisager.
Dans les SHEn impliquant d’autres tyrosine kinases (tel le transcrit ETV6-PDGFRB résultant de la translocation 5-12), l’imatinib mésylate est également indiqué mais à des posologies supérieures, semblables à celles utilisées dans la leucémie myéloïde chronique (soit 400 mg/j). Enfin, chez des patients ayant un SHE corticorésistant sans anomalie cytogénétique identifiée, mais ayant un « phénotype » compatible avec celui d’un syndrome myéloprolifératif, un test thérapeutique empirique avec l’imatinib mésylate peut également être proposé ponctuellement en raison de la possible implication d’autres tyrosine kinases non identifiables par les outils de biologie moléculaire actuels.10
Dans les situations autres que les SHEn, en cas d’inefficacité ou de corticodépendance à un seuil élevé, les traitements de deuxième ligne sont l’hydroxyurée et le peginterféron alpha-2a (PEG-IFNα), qui sont parfois même utilisés en association. Les principales limites du PEG-IFNα sont sa mauvaise tolérance au long cours (syndrome pseudo-grippal post-injection, troubles neuropsychiques), mais celle-ci peut être améliorée en débutant par des posologies faibles (par exemple 45 à 90 µg hebdomadaires de PEG-IFNα). Au contraire, l’hydroxyurée est généralement bien tolérée, bien que des toxicités (notamment hématologique ou cutanée) soient possibles.
Au cours des SHEr, des SHEl et des SHEi, les biothérapies ciblant l’IL-5 sont des options très séduisantes en raison de leur physiopathologie et du caractère « IL-5-dépendant » de l’éosinophilie. Dans une étude randomisée en double aveugle contre placebo chez 85 patients atteints de SHE F/P- corticodépendant, le mépolizumab (une IgG1 kappa murine « humanisée » qui neutralise l’IL-5 circulante) à la dose de 750 mg/mois par voie intraveineuse induisait une épargne cortisonique significative et un meilleur contrôle de l’hyperéosinophilie,11 et ce avec un profil de tolérance excellent. Malgré ces résultats prometteurs et ceux rapportés dans d’autres affections voisines (telles la polypose nasosinusienne et la granulomatose éosinophilique avec polyangéite), le mépolizumab n’a pas l’autorisation de mise sur le marché pour les SHE. Dans les SHE, une deuxième étude internationale prospective randomisée évaluant ce médicament (à la dose sous-cutanée de 300 mg mensuels) contre placebo est en cours. Plus récemment, une étude de phase II portant sur 20 patients ayant un SHE non lié à FIP1L1-PDGFRA et évaluant le benralizumab (un anticorps murin bloquant la sous-unité α du récepteur à l’IL-5) a montré des résultats prometteurs.12
Enfin, de manière plus anecdotique, de nombreux autres immunosuppresseurs (ciclosporine, méthotrexate, étoposide, vincristine) ont également été essayés au cours des SHE. Chez les patients multiréfractaires, l’alemtuzumab (anticorps monoclonal ciblant le CD-52), la cladribine et l’allogreffe de moelle sont les thérapeutiques de dernière ligne à envisager.
Quel est le pronostic ?
En 1975, la survie à 5 ans était seulement de 12 %.1 Plus récemment, dans une série française monocentrique de 49 patients suivis depuis 2000 et ayant pu bénéficier des traitements modernes (notamment inhibiteurs de tyrosine kinase et biothérapies ciblant l’IL-5), la survie était de 100 % à 5 ans, de 87 % à 10 ans et de 66 % à 15 ans. Bien que moins fréquente que dans les séries historiques, l’atteinte cardiaque est une des principales causes de mortalité, justifiant ainsi le dépistage régulier d’une éventuelle cardiopathie chez tout patient ayant une hyperéosinophilie chronique. Enfin, par analogie avec les autres hémopathies chroniques, il existe un risque d’acutisation en pathologie de haut grade : leucémie aiguë myéloblastique pour les patients ayant un SHEn ; lymphome T périphérique (notamment angio-immunoblastique) pour ceux ayant un SHEl.3
Hyperéosinophilie > 1.5G/L : toujours un signe d’alerte
La constatation d’une hyperéosinophilie sanguine, symptomatique ou non, ne doit jamais être négligée et doit toujours être un signe d’alerte pour le clinicien. Son origine « allergique » ne doit pas être retenue par excès, surtout si l’hyperéosinophilie dépasse 1,5 G/L. En cas de manifestation clinique (quelle qu’elle soit) rattachée à l’hyperéosinophilie, le diag-nostic de SHE doit être évoqué, et un bilan étiologique circonstancié ainsi qu’une prise en charge spécifique sont requises. Les causes de SHE sont multiples, mais l’interrogatoire combiné à un examen clinique minutieux et à des examens complémentaires orientés permettent souvent d’avancer dans le diagnostic. Néanmoins, on estime encore qu’environ la moitié des SHE restent « idiopathiques ».
La prise en charge des SHE a longtemps été fondée essentiellement sur la corticothérapie orale, l’hydroxyurée et l’IFNα. Si la meilleure compréhension des mécanismes moléculaires sous-jacents aux SHE et le développement de thérapies ciblées (tels les inhibiteurs de tyrosine kinase et les anticorps monoclonaux ciblant l’IL-5) ont révolutionné la prise en charge des patients, la place exacte de ces molécules et leurs modalités d’utilisation au long cours restent à mieux définir.
La prise en charge des SHE a longtemps été fondée essentiellement sur la corticothérapie orale, l’hydroxyurée et l’IFNα. Si la meilleure compréhension des mécanismes moléculaires sous-jacents aux SHE et le développement de thérapies ciblées (tels les inhibiteurs de tyrosine kinase et les anticorps monoclonaux ciblant l’IL-5) ont révolutionné la prise en charge des patients, la place exacte de ces molécules et leurs modalités d’utilisation au long cours restent à mieux définir.
Références
1. Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore) 1975;54:1‑27.
2. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012;130:607-612.e9.
3. Lefèvre G, Copin MC, Staumont-Sallé D, et al. ; French Eosinophil Network. The lymphoid variant of hypereosinophilic syndrome: study of 21 patients with CD3-CD4+ aberrant T-cell phenotype. Medicine (Baltimore) 2014;93:255‑66.
4. Ogbogu PU, Bochner BS, Butterfield JH, et al. Hypereosinophilic syndromes: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol 2009;124:1319-25.e3.
5. Leurs A, Chenivesse C, Lopez B, et al. C-reactive protein as a diagnostic tool in differential diagnosis of hypereosinophilic syndrome and antineutrophil cytoplasmic antibody-negative eosinophilic granulomatosis with polyangiitis. J Allergy Clin Immunol Pract 2019;7:1347-51.e3.
6. Groh M, Kahn JE, Ackermann F, et al. Orphanet urgences : syndromes hyperéosinophiliques. Orphanet, 2018. www.orpha.net http://bit.ly/2S53dz7
7. Klion AD. How I treat hypereosinophilic syndromes. Blood 2015;126:1069‑77.
8. Chen YY, Khoury P, Ware JM, et al. Marked and persistent eosinophilia in the absence of clinical manifestations. J Allergy Clin Immunol 2014;133:1195‑202.
9. Legrand F, Renneville A, Macintyre E, et al. The spectrum of FIP1L1-PDGFRA-associated chronic eosinophilic leukemia: new insights based on a survey of 44 cases. Medicine (Baltimore) 2013;92:e1-e9.
10. Khoury P, Desmond R, Pabon A, et al. Clinical features predict responsiveness to imatinib in platelet-derived growth factor receptor-alpha-negative hypereosinophilic syndrome. Allergy 2016;71:803‑10.
11. Rothenberg ME, Klion AD, Roufosse FE, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med 2008;358:1215‑28.
12. Kuang FL, Legrand F, Makiya M, et al. Benralizumab for PDGFRA-negative hypereosinophilic syndrome. N Engl J Med 2019;380:1336‑46.
2. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012;130:607-612.e9.
3. Lefèvre G, Copin MC, Staumont-Sallé D, et al. ; French Eosinophil Network. The lymphoid variant of hypereosinophilic syndrome: study of 21 patients with CD3-CD4+ aberrant T-cell phenotype. Medicine (Baltimore) 2014;93:255‑66.
4. Ogbogu PU, Bochner BS, Butterfield JH, et al. Hypereosinophilic syndromes: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol 2009;124:1319-25.e3.
5. Leurs A, Chenivesse C, Lopez B, et al. C-reactive protein as a diagnostic tool in differential diagnosis of hypereosinophilic syndrome and antineutrophil cytoplasmic antibody-negative eosinophilic granulomatosis with polyangiitis. J Allergy Clin Immunol Pract 2019;7:1347-51.e3.
6. Groh M, Kahn JE, Ackermann F, et al. Orphanet urgences : syndromes hyperéosinophiliques. Orphanet, 2018. www.orpha.net http://bit.ly/2S53dz7
7. Klion AD. How I treat hypereosinophilic syndromes. Blood 2015;126:1069‑77.
8. Chen YY, Khoury P, Ware JM, et al. Marked and persistent eosinophilia in the absence of clinical manifestations. J Allergy Clin Immunol 2014;133:1195‑202.
9. Legrand F, Renneville A, Macintyre E, et al. The spectrum of FIP1L1-PDGFRA-associated chronic eosinophilic leukemia: new insights based on a survey of 44 cases. Medicine (Baltimore) 2013;92:e1-e9.
10. Khoury P, Desmond R, Pabon A, et al. Clinical features predict responsiveness to imatinib in platelet-derived growth factor receptor-alpha-negative hypereosinophilic syndrome. Allergy 2016;71:803‑10.
11. Rothenberg ME, Klion AD, Roufosse FE, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med 2008;358:1215‑28.
12. Kuang FL, Legrand F, Makiya M, et al. Benralizumab for PDGFRA-negative hypereosinophilic syndrome. N Engl J Med 2019;380:1336‑46.
Dans cet article


Résumé
Les syndromes hyperéosinophiliques sont définis par l’association d’une hyperéosinophilie sanguine supérieure ou égale à 1,5 G/L d’évolution chronique (> 1 mois) à des dommages tissulaires (quels qu’ils soient) en rapport avec l’infiltration éosinophilique. Il s’agit d’une entité hétérogène qui comprend notamment les syndromes hyperéosinophiliques néoplasiques « clonaux » (SHEN) [dont la leucémie chronique à éosinophiles liée à la délétion FIP1L1-PDGFRA et les éosinophilies associées aux autres syndromes myéloprolifératifs et myélodysplasiques] et les syndromes hyperéosinophiliques réactionnels (SHER, entité hétérogène regroupant l’ensemble des situations (infections parasitaires, prise médicamenteuse, maladies inflammatoires ou néoplasiques) responsables de la production de cytokines Th2 conduisant à une hyperéosinophilie non clonale. Parmi les SHER, on distingue les SHE lymphoïdes (SHEL), où la production d’interleukine 5 (IL-5) est liée à la présence d’une lymphoprolifération T de bas grade de phénotype aberrant (généralement CD3-CD4+). Malgré un bilan causal exhaustif large, on estime qu’environ 50 % des SHE restent d’origine indéterminée. Les manifestations cliniques sont diverses et les atteintes dermatologiques, respiratoires et digestives sont les plus fréquentes. Le pronostic à long terme est surtout corrélé à l’atteinte cardiaque et, pour les SHEN et les SHEL, au risque d’acutisation en pathologie maligne de haut grade (leucémie aiguë myéloblastique et lymphome T périphérique respectivement). La prise en charge des SHEN repose sur les inhibiteurs de tyrosine kinase, notamment l’imatinib mésylate. Pour les SHER (y compris les SHEL), la corticothérapie est généralement efficace, et les thérapeutiques de deuxième ligne sont l’hydroxyurée et le peginterféron alpha-2a. Les biothérapies ciblant l’IL-5 sont très prometteuses (hors SHEN) mais leur utilisation est pour l’instant limitée aux essais thérapeutiques et à un protocole d’usage compassionnel pour les patients les plus sévères et réfractaires aux thérapeutiques de première ligne.