Définir les éléments cliniques et de diagnostic d’une maladie de Vaquez, d’une thrombocytémie primitive, d’une leucémie myéloïde chronique.
Généralités
Définition et physiopathologie
Ils sont caractérisés par l’hyperplasie d’une ou plusieurs lignées myéloïdes au niveau médullaire, sans blocage de maturation contrairement aux leucémies aiguës. Ceci se traduit sur l’hémogramme par une augmentation d’un ou plusieurs types de cellules circulantes, morphologiquement normales.
Ils sont la conséquence de l’acquisition dans une cellule souche hématopoïétique d’une anomalie génétique somatique entraînant une activation anormale de la signalisation intracellulaire et une dérégulation de la prolifération cellulaire devenant indépendante des facteurs de croissance. Les causes de survenue de cette anomalie génétique ne sont pas connues.
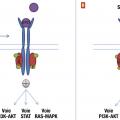
Les anomalies oncogéniques les mieux identifiées et les plus fréquentes dans les syndromes myéloprolifératifs impliquent des protéines à activité tyrosine kinase : BCR-ABL1 dans la leucémie myéloïde chronique (LMC), JAK2 dans les 3 principaux syndromes myéloprolifératifs classiques non LMC (
Classification
- la polyglobulie de Vaquez : prolifération prédominant sur la lignée érythrocytaire ;
- la thrombocytémie essentielle : prolifération prédominant sur la lignée mégacaryocytaire ;
- la myélofibrose primitive : prolifération pouvant toucher les 3 lignées myéloïdes associée à de la fibrose médullaire.
Circonstances diagnostiques
Évolution
À court et moyen terme, leur principal risque est thrombotique (thromboses veineuses comme artérielles). Ces complications sont liées à l’hyperviscosité sanguine secondaire à l’augmentation de la masse sanguine dans les polyglobulies, mais aussi à des propriétés particulières d’adhésivité des leucocytes et des plaquettes, et possiblement à un état pro-inflammatoire. Dans la LMC, on peut noter également un risque vasculaire artériel induit par certains traitements inhibiteurs de kinases.
À long terme, les polyglobulies de Vaquez et thrombocytémies essentielles sont à risque d’évolution vers une myélofibrose secondaire. Tous les syndromes myéloprolifératifs sont également à risque d’évolution vers un syndrome myélodysplasique ou une leucémie aiguë (le plus souvent myéloblastique), de pronostic très sombre.
Leucémie myéloïde chronique (LMC)
Définition et physiopathologie
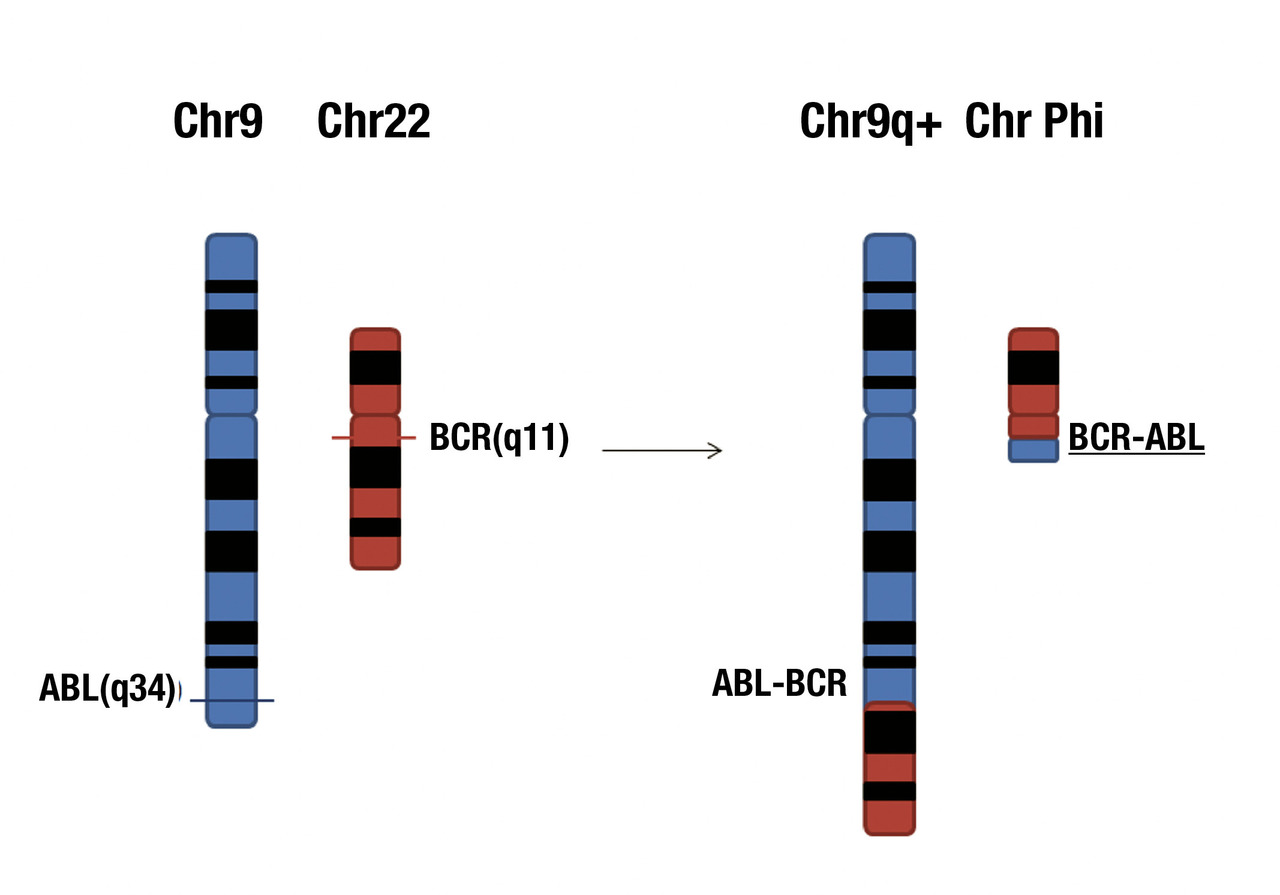
La translocation t(9;22) est une translocation réciproque et équilibrée (
À l’échelle moléculaire, cette translocation aboutit à la formation d’un gène et d’un transcrit de fusion entre les gènes BCR (sur le chromosome 22) et ABL1 (sur le chromosome 9).
Selon le point de cassure, il existe différents transcrits. Les transcrits b3a2 et b2a2 sont les transcrits les plus fréquents, appelés M BCR-ABL1 (pour Major BCR-ABL1), ils représentent à eux deux 85 % des leucémies myéloïdes chroniques. Le transcrit e1a2 (mBCR-ABL1, pour minor BCR-ABL1) représente 0,4 % des cas, il est retrouvé également dans deux tiers des leucémies aiguës lymphoblastiques.
Le gène ABL1 code une protéine tyrosine kinase cytoplasmique qui interagit avec de nombreuses voies de signalisation. Sa fusion avec le gène BCR engendre l’activation constitutive de la kinase. En découlent : une prolifération excessive de la lignée granuleuse, une diminution de l’apoptose, une perte d’adhérence cellulaire.
Épidémiologie
Dans la majorité des cas, son étiologie est inconnue. Mais il peut être identifié un facteur causal dans moins de 5 % des cas : exposition chronique aux benzènes, aux radiations ionisantes.
Circonstances diagnostiques
Diagnostic positif
- une hyperleucocytose franche, globules blancs > 100 G/L dans la moitié des cas ; à polynucléaires neutrophiles (40 à 60 % de la formule leucocytaire), avec excès de polynucléaires basophiles quasi constant (jusqu’à 10-15 % de la formule) et myélémie (30 à 60 % de la formule). La myélémie est équilibrée avec surtout des métamyélocytes et des myélocytes, et quelques promyélocytes. Les blastes circulants et myéloblastes sont < 5 % (sinon envisager une phase accélérée). Il peut exister un petit excès de polynucléaires éosinophiles, parfois jusqu’à 20 % de la formule. Le pourcentage de lymphocytes et monocytes est normal en général. On note une possible hypermonocytose, associée à un transcrit de fusion particulier (e1a2) ;
- une thrombocytose modérée fréquente (50 % des cas). À noter qu’elle peut, rarement, être prédominante ou isolée ;
- une anémie absente ou modérée.
- RT-PCR en biologie moléculaire dans le sang, à la recherche du transcrit ARNm BCR-ABL1 ;
- caryotype des cellules de la moelle obtenues par ponction médullaire, à la recherche de la translocation t(9 ;22) ;
- FISH (hybridation in situ) sur ADN du noyau avec des sondes complémentaires des gènes ABL1 et BCR marquées par des fluorochromes de couleurs différentes.
Autres éléments de la démarche diagnostique et préthérapeutique
Un bilan médullaire est indispensable également (myélogramme, caryotype médullaire ± FISH, transcrit BCR-ABL) :
- le myélogramme permet avant tout de vérifier l’absence d’accélération ou transformation : absence d’excès de blastes. On retrouve une hyperplasie granuleuse (80-90 %), une maturation normale, une éosinophilie et basophilie médullaire ;
- le caryotype, outre le chromosome Philadelphie, permet la recherche d’anomalies complexes ou additionnelles qui signeraient elles aussi une accélération ou une transformation (perte du Y, trisomie du 8, duplication du chromosome Philadelphie, isochromosome 17q).
Un bilan métabolique doit être réalisé : uricémie, fonction hépatique, fonction rénale, LDH.
Diagnostic différentiel
Face à une hyperleucocytose modérée, on évoque les causes infectieuses, inflammatoires dont les cancers métastatiques, ou iatrogènes (corticoïdes, adrénaline), les régénérations médullaires (post-aplasie ou post-saignement), le tabac.
Face à une hyperleucocytose avec myélémie déséquilibrée avec un excès de cellules immatures et/ou des éléments érythroblastiques circulants, on pourra évoquer un autre syndrome myéloprolifératif (myélofibrose primitive, syndrome myéloprolifératif-myélodysplasique). Mais on ne retrouvera pas de transcrit BCR-ABL.
Face à une thrombocytose prédominante, on évoquera une thrombocytose réactionnelle ou une thrombocytémie essentielle.
Complications
Complications hématologiques
Ce sont :- les hémorragies liées à la thrombopathie ;
- les thromboses liées à l’hyperleucocytose et la thrombocytose (infarctus splénique, priapisme, syndrome de Budd-Chiari) ;
- la leucostase cérébrale (ralentissement psychomoteur jusqu’au coma), pulmonaire (détresse respiratoire).
Complications métaboliques
Ce sont :- les complications de l’hyperuricémie (crise de goutte) ;
- l'insuffisance rénale ;
Évolution
La phase chronique durait en moyenne de 3 à 5 ans (en l’absence de traitement).
La phase accélérée, inconstante, dure en moyenne de 12 à 18 mois. Cette phase accélérée est caractérisée par une augmentation du volume de la rate, l’apparition éventuelle de signes généraux (amaigrissement, sueurs nocturnes, fièvre sans infection) et de douleurs osseuses. Au niveau biologique, on note une augmentation de la leucocytose, une basophilie (à plus de 20 %), l’apparition d’une thrombopénie. On diagnostique cette phase accélérée lorsqu’on trouve un pourcentage de blastes entre 10 et 19 % dans le sang et/ou la moelle. La résistance aux inhibiteurs de kinase doit également faire rechercher un passage en phase accélérée.
La phase de transformation aiguë correspond à l’évolution vers une leucémie aiguë myéloblastique dans deux tiers des cas, ou lymphoblastique B dans un tiers des cas. Elle est de pronostic très défavorable, avec une survie médiane de 3 mois en l’absence de traitement. Elle se caractérise par l’aggravation de la splénomégalie, des signes généraux, des douleurs osseuses et l’apparition possible de localisations cutanées, pulmonaires ou neurologiques. Au niveau biologique, on peut noter l’augmentation des éosinophiles et basophiles, l’apparition d’anémie ou thrombopénie, enfin la disparition de la myélémie avec apparition de blastes. Au myélogramme, on note plus de 20 % de blastes. Au caryotype, on note fréquemment l’apparition d’anomalies supplémentaires.
Principes de traitement
Inhibiteurs de tyrosine kinase
Le traitement de la leucémie myéloïde chronique a connu une véritable révolution à l’arrivée des médicaments ciblés de type inhibiteurs de tyrosine kinase au début des années 2000. Cette maladie à haut risque de transformation, alors première indication d’allogreffe, est devenue une maladie chronique, contrôlée par des traitements per os administrés au long cours :- imatinib (Glivec), inhibiteur de tyrosine kinase de 1re génération, posologie de 400 mg/j en phase chronique, largement utilisé en 1re ligne de traitement ;
- dasatinib et nilotinib, inhibiteurs de tyrosine kinase de 2e génération, utilisés en 1re ou 2e ligne. Ils sont potentiellement plus puissants mais aussi pourvoyeurs de plus d’effets secondaires potentiels ;
- bosutunib et ponatinib, inhibiteurs de tyrosine kinase de 3e génération, au-delà de la 1re ligne, voire 2e ligne, utilisés dans des indications plus spécifiques.
Suivi de la maladie et de la réponse au traitement
Une surveillance régulière des patients est indispensable, et repose sur :- un examen clinique régulier ;
- l’hémogramme, fréquent jusqu’à normalisation, puis plus espacé ;
- le caryotype médullaire répété tous les 3 à 6 mois jusqu’à disparition du chromosome Philadelphie ;
- le suivi du transcrit moléculaire BCR-ABL1 sanguin, tous les 3 mois la ou les première(s) année(s), puis tous les 6 mois à vie (y compris s’il devient indétectable) ;
- l’évaluation régulière de l’observance, qui est un enjeu majeur de la réponse lors des traitements au long cours.
Place de l’allogreffe de cellules hématopoïétique
Elle n’a plus sa place en première intention dans le traitement des formes chroniques depuis l’avènement des inhibiteurs de tyrosine kinase. Elle reste indiquée dans les cas de résistance aux inhibiteurs de tyrosine kinase, de transformation aiguë, ou chez les enfants.Avant l’arrivée des inhibiteurs de tyrosine kinase qui ont révolutionné le pronostic de la leucémie myéloïde chronique, l’histoire naturelle de la maladie était l’évolution vers la leucémie aiguë.
Sous inhibiteur de tyrosine kinase, la leucémie myéloïde chronique est devenue une maladie chronique d’excellent pronostic
Polyglobulie de Vaquez
Définition et physiopathologie
La polyglobulie de Vaquez est une polyglobulie primitive, c’est-à-dire que les précurseurs hématopoïétiques porteurs de la mutation de JAK2 prolifèrent en excès sans nécessité de stimulation par l’érythropoïétine (EPO). Ceci en opposition aux polyglobulies secondaires au cours desquelles l’hyperproduction de cellules érythroïdes est due à une production excessive d’EPO. La mutation intervenant dans une cellule souche hématopoïétique, il existe une hyperplasie myéloïde médullaire globale, qui prédomine cependant sur la lignée érythroblastique.
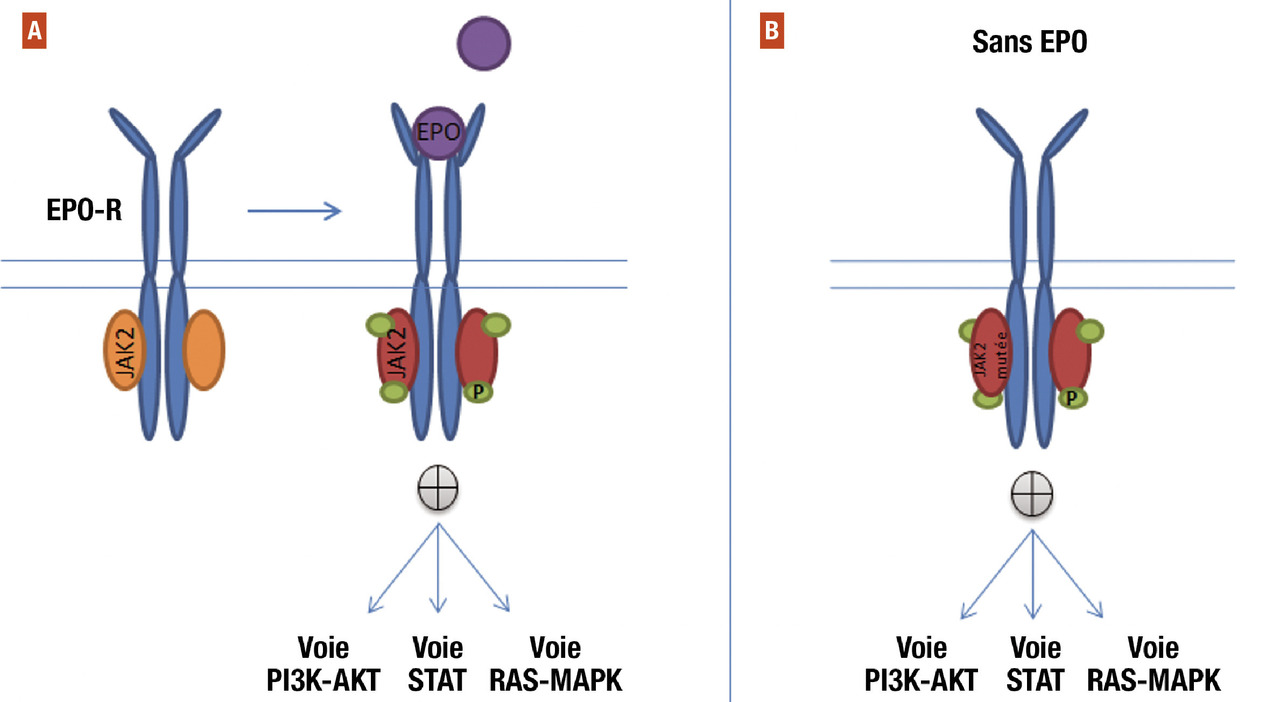
La mutation de JAK2 retrouvée dans 95 % des cas est la mutation JAK2 V617F. C’est une mutation activatrice. Il en résulte une protéine JAK2 mutée qui présente une activité tyrosine kinase constitutive (
La protéine JAK2 normale est associée au récepteur de l’EPO. La fixation de l’EPO sur le récepteur entraîne des changements de conformation spatiale du récepteur qui activent JAK2. JAK2 activée phosphoryle le récepteur lui-même puis des protéines intracytoplasmiques (seconds messagers), ce qui enclenche des voies de signalisation intracellulaire de réponse à l’EPO et de prolifération cellulaire. Dans la polyglobulie de Vaquez, la protéine JAK2 mutée, constitutivement activée, n’a pas besoin du signal de l’EPO pour activer la signalisation cellulaire. Les cellules porteuses de la mutation deviennent donc capables de proliférer sans EPO.
Il est à noter que cette anomalie (mutation ponctuelle) n’est pas visible à l’échelle cytogénétique et qu’il n’y a par ailleurs pas d’anomalie cytogénétique spécifique de la polyglobulie de Vaquez. Le caryotype médullaire est donc normal dans la majorité des cas.
Épidémiologie
Circonstances diagnostiques
- découverte fortuite sur hémogramme réalisé lors d’un bilan ;
- érythrose cutanéomuqueuse (le plus souvent constatée sur le visage et les mains) ou conjonctivale d’apparition progressive ;
- symptômes microvasculaires liés à l’hyperviscosité obstruant les capillaires les plus fins : céphalées, vertiges, acouphènes, scotomes, érythromélalgie ;
- complication vasculaire inaugurale : ischémique (thrombose veineuse, notamment d’un territoire rare comme thrombose porte ou syndrome de Budd-Chiari, thrombose veineuse cérébrale, mais aussi dans les territoires plus classiques, thrombose artérielle dont infarctus du myocarde ou accident vasculaire cérébral), hémorragique plus rarement (épistaxis notamment) ;
- symptôme évocateur de syndrome myéloprolifératif : prurit aquagénique, splénomégalie.
Diagnostic positif
Hémogramme
L'hémogramme retrouve :- une augmentation du taux d’hémoglobine : Hb > 16,5 g/dL chez l’homme, Hb > 16 g/dL chez la femme.
- une augmentation du taux d’hématocrite : Ht > 49 % chez l’homme, Ht > 48 % chez la femme.
- une thrombocytose inconstante ;
- une hyperleucocytose modérée inconstante, prédominant sur les polynucléaires neutrophiles.
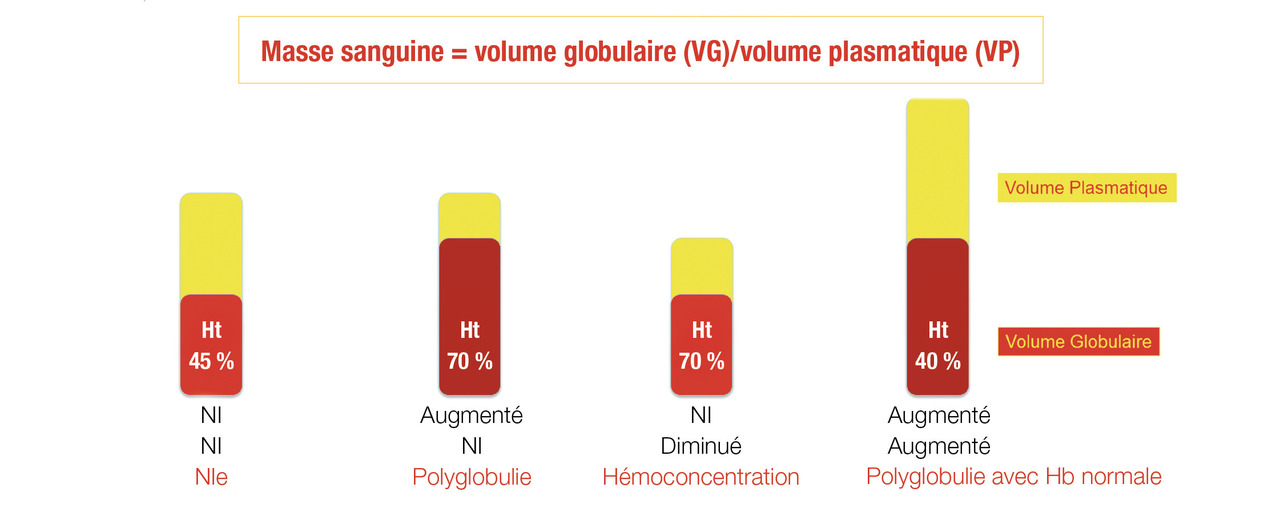
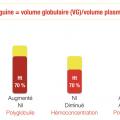
Mesure isotopique de la masse sanguine
Les taux d’hémoglobine et d’hématocrite étant dépendants de la dilution plasmatique, en cas de doute, on peut réaliser une mesure isotopique de la masse sanguine, qui permet de formellement diagnostiquer une polyglobulie vraie lorsque le volume globulaire total, mesuré par des hématies marquées au 99Tc, est augmenté de plus de 25 % par rapport à la valeur théorique calculée pour le patient (Mutations du gène JAK2
La mutation JAK2 V617F est retrouvée dans plus de 95 % des cas de Vaquez. Une mutation différente, dans l’exon 12 de JAK2 est trouvée dans environ 2 % des cas qui n’ont pas la mutation JAK2 V617. Au total donc, une mutation de JAK2 est présente dans plus de 97 % des cas, et son absence doit faire reconsidérer le diagnostic de polyglobulie de Vaquez .L’examen est facile à réaliser sur un bilan sanguin (par PCR) et est donc maintenant réalisé en 1re intention.
La mutation JAK2 V617F n’est cependant pas pathognomonique de la polyglobulie de Vaquez puisqu’elle est retrouvée dans environ 50 % des thrombocytémies essentielles et myélofibroses.
Biopsie ostéomédullaire
Elle trouve une moelle hypercellulaire pour l’âge, avec hyperplasie des 3 lignées myéloïdes, prédominant sur la lignée érythroïde.Elle permet d’évaluer le degré de fibrose potentiellement présent (une fibrose médullaire de grade > 1 fait basculer vers un diagnostic de myélofibrose).
Dosage d’EPO sérique (réalisé avant toute saignée)
Il est diminué ou normal en cas de polyglobulie de Vaquez.À l’inverse, il est normal ou augmenté en cas de polyglobulie secondaire. Il existe des zones de recouvrement qui rendent parfois l’interprétation du résultat délicate.
Culture des progéniteurs érythroblastiques in vitro
Cet examen in vitro à la recherche d’une « pousse spontanée » des progéniteurs érythroblastiques n’est quasiment plus pratiquée.Critères de l’Organisation mondiale de la santé (OMS) 2016 pour un diagnostic de polyglobulie de Vaquez
Critères majeurs
ou hématocrite > 49 % (homme) ou > 48 % (femme),
ou augmentation de la masse sanguine (> 25 % au-delà de la valeur normale attendue).
2) Biopsie ostéomédullaire montrant une hypercellularité pour l’âge avec prolifération excessive des 3 lignées myéloïdes (panmyélose), incluant une prolifération de mégacaryocytes polymorphes et matures (avec des tailles cellulaires différentes).
3) Présence d’une mutation JAK2V617F ou JAK2 exon 12.
Critère mineur
Le diagnostic de polyglobulie de Vaquez nécessite soit l’ensemble des 3 critères majeurs, soit les 2 premiers et le critère mineur.
Le critère 2 (biopsie ostéomédullaire) n’est pas indispensable en cas d’érythrocytose absolue (Hb > 18,5 g/dL [ou Ht > 55,5 %] chez l’homme ou > 16,5 g/dL [Ht > 49,5 %] chez la femme) si le critère majeur 3 et le critère mineur sont tous deux présents. Cependant, une fibrose médullaire est présente chez 10-20 % des patients au diagnostic et ne peut être détectée que par biopsie ostéomédullaire (critère de risque d’évolution plus rapide vers une myélofibrose post-polyglobulie de Vaquez ).
Démarche diagnostique en pratique
À l’examen clinique dans un 1er temps, on recherche des signes en faveur d’une hémoconcentration (déshydratation, prise de médicaments diurétiques), des signes particulièrement évocateurs de Vaquez (prurit aquagénique, splénomégalie, érythrose faciale, signes microvasculaires, signes de complication thrombotique inaugurale), des signes évocateurs de polyglobulie secondaire (signes d’insuffisance respiratoire, signes évocateurs d’apnées du sommeil, consommation de chicha, notion de tumeur rénale, transplantation rénale, syndrome cérébelleux).On demandera dans le sang une recherche de mutation JAK2 V617F et un dosage d’EPO en premier lieu.
Si la mutation JAK2V617F est retrouvée, on discutera la réalisation ou non de la biopsie ostéomédullaire pour compléter la démarche diagnostique en fonction des chiffres de l’hémogramme.
Si la mutation JAK2 V617F est négative avec EPO basse, on recherchera une mutation de l’exon 12 de JAK2, puis on réalisera une biopsie ostéomédullaire.
Si la mutation JAK2 V617F est négative avec EPO normale ou élevée, on cherchera à affirmer la polyglobulie vraie, avec réalisation d’une masse sanguine isotopique. Si celle-ci confirme la polyglobulie, on se dirigera vers une recherche de cause de polyglobulie secondaire.
Autres éléments de la démarche diagnostique et préthérapeutique
Un bilan d’hémostase est indiqué, en particulier en cas de thrombocytose, pour évaluer le risque hémorragique : au-delà d’une évaluation du temps de prothrombine (TP) et ratio de temps de céphaline activé (TCA ratio), on pourra être amené à rechercher une maladie de Willebrand acquise par les dosages de facteur VIII, d’antigène et d’activité du facteur Willebrand.
Bilan biologique rénal, hépatique, dosage d’acide urique, LDH font partie du bilan initial d’une polyglobulie de Vaquez.
Le bilan des facteurs de risque cardiovasculaire (tabagisme, mesure de la tension artérielle, dosage de la glycémie à jeun, des triglycérides, du cholestérol total, HDL, LDL) est également indispensable après un diagnostic de polyglobulie de Vaquez, qui doit être considérée comme un facteur de risque cardiovasculaire supplémentaire. Il convient chez les patients avec une polyglobulie de Vaquez d’opérer un strict contrôle de l’ensemble des facteurs de risque cardiovasculaires au long cours.
Diagnostic différentiel
Fausses polyglobulies
Hémoconcentrations
Déshydratation, prise de diurétiques, brûlures étendues.
Syndromes thalassémiques hétérozygotes
Le nombre d’hématies n’est pas un critère de diagnostic de polyglobulie.
Syndrome de Gaisböck
Polyglobulies secondaires acquises
Hypoxie centrale
Hypoxie locale
Production anormale d’EPO par une tumeur
Médicamenteuse
Polyglobulies constitutionnelles
Elles sont extrêmement rares, il existe en général une notion d’antécédent familial de polyglobulie.On peut citer les polyglobulies constitutionnelles primitives liées à des mutations du gène du récepteur de l’EPO. Il peut aussi s’agir de polyglobulies constitutionnelles secondaires à : une hémoglobinopathie rendant l’hémoglobine hyperaffine pour l’oxygène, des mutations de gènes impliqués dans la voie de la réponse à l’hypoxie (comme VHL, PHD2…).
Autres syndromes myéloprolifératifs
On retrouve la mutation JAK2 V617F dans 50 % des thrombocytémies essentielles et des myélofibroses, mais dans ces dernières il n’y a pas de polyglobulie.Complications et évolution
Complications de l’hyperuricémie
Thromboses artérielles et veineuses
Il faut considérer la polyglobulie de Vaquez comme un facteur de risque cardiovasculaire à part entière, qui majore le risque d’athérosclérose et notamment d’accident coronarien, d’accident vasculaire cérébral.
Les thromboses veineuses pouvant compliquer la polyglobulie de Vaquez ont potentiellement des sites particuliers : thromboses splanchniques (thrombose porte, syndrome de Budd-Chiari), thromboses veineuses cérébrales. Le risque thrombotique post- opératoire est également augmenté et prolongé.
Hémorragies
Une source de retard au diagnostic est le saignement digestif occulte occasionnant une carence martiale masquant la polyglobulie.
Risque évolutif à long terme
Pronostic
Le risque thrombotique est diminué par un bon contrôle de la pathologie par les traitements cytoréducteurs mais reste supérieur à celui de la population générale. Le risque hémorragique est en revanche augmenté par l’antiagrégation ou l’anticoagulation. Aucun traitement n’a fait preuve d’une efficacité pour diminuer le risque de transformation.
L’espérance de vie médiane se situe entre 15 et 20 ans. Elle semble diminuée de quelques années par rapport à la population générale dans les études internationales, un phénomène qui n’a pas été confirmé dans une étude française avec un suivi prospectif médian de plus de 16 ans.
Principe de traitement
Antiagrégation et anticoagulation
L’aspirine à dose antiagrégante (100 mg × 1 ou × 2/j) a montré son efficacité dans la prévention des thromboses chez les patients avec polyglobulie de Vaquez et est systématiquement prescrite sauf contre-indication absolue.Les anticoagulants n’ont leur place qu’en cas de survenue de thrombose veineuse. Le traitement est alors à envisager au long cours, sauf contre-indication.
Saignées
Elles sont à réaliser en urgence en cas d’hématocrite > 60 % ou de signes d’hyperviscosité. Elles n’ont pas de contre-indication mais sont à réaliser avec prudence chez le sujet âgé, en surveillant la tolérance hémodynamique.On réalise une ponction veineuse de 300 à 400 mL par saignée. Elles peuvent être répétées 2 ou 3 fois par semaine initialement. L’objectif est de ramener l’hématocrite en dessous de 45 %. Dans certains cas d’hématocrite très élevé, on peut réaliser une érythraphérèse, ayant l’avantage en une séance de ramener l’hématocrite dans la zone souhaitée.
Les saignées ont une action immédiate sur le risque thrombotique en réduisant rapidement le volume sanguin et l’hématocrite. Quand elles sont faites de manière chronique, elles ont une action retardée par la carence martiale qu’elles induisent et qui freine l’érythropoïèse. Il est ainsi important de respecter la carence martiale induite, il faut prévenir le patient et ses différents médecins de ne pas donner de fer, sauf cas particuliers symptomatiques.
Elles induisent en revanche potentiellement une thrombocytose, et la carence martiale peut devenir symptomatique en elle-même, de sorte que la stratégie de saignées itératives est parfois difficile à maintenir comme traitement de fond.
Médicaments cytoréducteurs
Une cytoréduction médicamenteuse est indiquée chez les patients à haut risque thrombotique : patients de plus de 60 ans, et/ou ayant un antécédent de thrombose, et/ou en cas de thrombocytose supérieure à 1 000 G/L. Elle peut également être débutée en cas d’intolérance des saignées au long cours, d’apparition d’hyperleucocytose ou d’une splénomégalie symptomatique.Ils ont fait la preuve de leur efficacité pour réduire le risque thrombotique ou hémorragique, mais aucun n’a pour l’instant fait preuve d’une efficacité à réduire le risque de transformation en myélofibrose ou leucémie aiguë à long terme. Certains posent même le problème d’un potentiel leucémogène.
Deux médicaments ont l’indication en 1re ligne : hydroxyurée et ropéginterféron (AMM européenne obtenue en mars 2019, mais pas encore disponible sur le marché en France lors de la rédaction de cet article).
Deux médicaments ont l’indication 2e ligne : ruxolitinib et pipobroman.
Enfin, le peginterféron alfa-2a n’a pas l’autorisation de mise sur le marché dans cette indication, mais est utilisé (recommandations internationales des sociétés savantes) en particulier chez les patients jeunes et chez les femmes avec désir de grossesse et pendant la grossesse. L’avantage des interférons est l’absence de risque leucémogène.
Hydroxyurée : gélules de 500 mg, dose moyenne 2 gel/j, adaptation de la dose au contrôle des NFS (rapproché les premières semaines de traitement). On note qu’il entraîne une macrocytose de par son mécanisme d’action (antimétabolite) sans conséquence par elle-même. Ses principaux effets secondaires, en dehors de la myélosuppression, sont cutanés (xérose, ulcères de jambes, qui sont une indication formelle d’arrêt du traitement, sur-risque de carcinomes cutanés au long cours).
Ropéginterféron alpha-2b : injections sous-cutanées bimensuelles en phase d’attaque, puis mensuelles, dose initiale 100 µg/15 jours, adaptation de la dose aux contrôles des numérations formule sanguine. Ses principaux effets secondaires en dehors de la myélosuppression sont la fatigue, les perturbations du bilan hépatique, les troubles de l’humeur, le déclenchement de pathologies auto-immunes ou inflammatoires (principalement l’hypothyroïdie). Il sera le traitement de choix chez le jeune patient en l’absence de contre-indication.
Ruxolitinib : comprimés de 5, 10, 15 ou 20 mg, la dose initiale est de 10 mg × 2/j. C’est un inhibiteur de JAK2. Il a obtenu l’AMM en cas d’échec ou d’intolérance de l’hydroxyurée. Sa tolérance clinique est en général excellente, il est très efficace sur les symptômes de polyglobulie de Vaquez : il diminue l’asthénie, est très efficace sur le prurit, et réduit la taille de la rate. On peut noter dans les effets secondaires : prise de poids, désordres digestifs, céphalées, hypertension, risque infectieux (en particulier réactivations herpétiques). Il semble également majorer le risque de carcinomes cutanés, notamment chez les patients ayant des antécédents de lésion cutanée et exposés à l’hyroxyurée pendant une longue période.
Pipobroman : comprimés de 25 mg. Son profil de tolérance clinique est excellent. Il présente en revanche un potentiel leucémogène (proche des alkylants) qui nous le fera réserver en échec des autres traitements ou chez les patients âgés.
Thrombocytémie essentielle
Définition et physiopathologie
La mutation de JAK2 V617F est trouvée chez 50% des patients, la protéine JAK2 étant impliquée dans la signalisation du récepteur de la thrombopoïétine. Des mutations dans d’autres gènes peuvent être impliquées, chez des patients non porteurs de la mutation JAK2 V617F : mutations de CALR (gène codant pour la calréticuline) dans 25-30 % des cas, mutation de MPL (gène du récepteur de la thrombopoïétine) dans 5 % des cas. Il reste encore cependant environ 15-20 % des cas pour lesquels on ne retrouve pas de mutation dans un de ces 3 gènes, on parle alors de thrombocytémie essentielle « triple négative »
Il n’existe pas d’anomalie cytogénétique spécifique de la thrombocytémie essentielle, et le caryotype médullaire est normal dans la majorité des cas.
Épidémiologie
Circonstances diagnostiques
Parfois, la thrombocytémie essentielle est découverte sur l’exploration de signes cliniques, en particulier vasculaires :
- érythromélalgies (signe fonctionnel très évocateur ; violentes douleurs des pieds et des mains, à type de brûlures, avec érythème simultané et éventuel œdème ; elles sont liées à des occlusions de la microcirculation artérielle). Ce symptôme est très sensible à l’aspirine, et doit disparaître en quelques minutes après la prise (test diagnostique) ;
- cortège de symptômes microvasculaires (céphalées, vertiges, phosphènes, scotomes, acouphènes…) ;
- complication vasculaire inaugurale : ischémique (thrombose veineuse, notamment d’un territoire particulier comme thrombose porte ou syndrome de Budd-Chiari, thrombose veineuse cérébrale, mais aussi dans les territoires plus classiques, thrombose artérielle dont infarctus du myocarde ou accident vasculaire cérébral), hémorragique plus rarement (épistaxis notamment) ;
- splénomégalie (moins de 50 % des cas, en général modérée).
Diagnostic positif
Hémogramme
Il montre une thrombocytose > 450 G/L, en général isolée, persistant sur plusieurs hémogrammes. Il peut exister une discrète hyperleucocytose à polynucléaires neutrophiles. Il n’y a pas d’anomalie de l’hémoglobine ou de l’hématocrite.En premier lieu face à une thrombocytose, il faut s’assurer de son caractère chronique, puis éliminer une thrombocytose réactionnelle, en particulier à une carence martiale ou à un syndrome inflammatoire (cf. diagnostics différentiels).
Une fois ces causes éliminées, on recherche sur sang les mutations JAK2 V617F, puis de CALR, puis de MPL.
Le diagnostic positif de thrombocytémie essentielle nécessite la réalisation systématique d’une biopsie ostéomédullaire depuis la mise à jour de la classification OMS en 2016. En effet, ses caractéristiques sont un des critères majeurs de la classification. Elle permet en particulier d’éliminer une myélofibrose préfibrotique ou une polyglobulie masquée, au cours desquelles la clinique et la NFS sont possiblement similaires.
Critères de l’OMS 2016 pour un diagnostic de thrombocytémie essentielle
Critères majeurs
2) Biopsie ostéomédullaire montrant une prolifération principalement de la lignée mégacaryocytaire, avec un nombre augmenté de grands mégacaryocytes matures, au noyau hyperlobé. Pas d’augmentation significative ou de hiatus dans la lignée granulocytaire ou érythroblastique, et absence d’augmentation (ou très mineure = grade 1) des fibres de réticuline.
3) Pas de critères de l’OMS pour une leucémie myéloïde chronique, une polyglobulie de Vaquez, une myélofibrose primitive, un syndrome myélodysplasique ou une autre néoplasie myéloïde.
4) Présence d’une mutation de JAK2, CALR ou MPL.
Critère mineur
Le diagnostic de thrombocytémie essentielle requiert les 4 critères majeurs ou les 3 premiers critères majeurs et le critère mineur.
Autres éléments de la démarche diagnostique et préthérapeutique
Il sera parfois nécessaire de réaliser un myélogramme avec coloration de Perls et caryotype médullaire en cas de suspicion de syndrome myélodysplasique.
Tout comme pour la plyglobulie de Vaquez, on réalisera également :
- une échographie abdominale pour évaluer l’éventuelle splénomégalie ;
- un bilan d’hémostase minimal avec TP et TCAr et la recherche éventuelle de syndrome de Willebrand acquis (FVIII, antigène et activité du facteur Willebrand) ;
- un bilan biologique rénal, hépatique, dosage d’acide urique, LDH ;
- un bilan des autres facteurs de risque cardiovasculaire (tabagisme, mesure de la tension artérielle, dosage de la glycémie à jeun, des triglycérides, du cholestérol total, HDL, LDL), puisque la thrombocytémie essentielle doit également être considérée comme un facteur de risque cardiovasculaire à part entière.
Diagnostic différentiel
Thrombocytoses réactionnelles
Elles constituent largement les causes les plus fréquentes de thrombocytose et sont à éliminer systématiquement en 1re intention.On peut citer les causes de thrombocytoses aiguës transitoires, facilement identifiées par le contexte clinique : régénération médullaire, sortie d’aplasie, infection aiguë, état inflammatoire aigu.
Les causes de thrombocytoses réactionnelles chroniques occasionnent rarement des thrombocytoses supérieures à 800 G/L. Elles sont constituées de :
Carence martiale
On recherche un contexte hémorragique chronique (ménorragies hémorragiques, méléna ou rectorragies, traitement anticoagulant…) ; à la biologie, on pourra noter une anémie microcytaire ou une microcytose isolée, une ferritinémie diminuée.Syndrome inflammatoire
On recherche des antécédents de maladie inflammatoire, un contexte infectieux, des éléments orientant vers un cancer ; à la biologie, on cherchera une CRP augmentée.Splénectomie (ou asplénie fonctionnelle plus rarement)
En post-splénectomie immédiat, la thrombocytose peut être majeure (au-delà de 1 000 G/L), puis elle redescend en général après quelques semaines, avec un chiffre de plaquettes qui peut se normaliser ou rester discrètement augmenté.Autres syndromes myéloprolifératifs
La leucémie myéloïde chronique peut se manifester par une thrombocytose au premier plan, isolée ou accompagnée d’une hyperleucocytose et d’une myélémie. On retrouvera alors un transcrit BCR-ABL.La maladie de Vaquez peut parfois se présenter avec une thrombocytose prédominante, en particulier en cas de carence martiale associée qui masque la polyglobulie et majore la thrombocytose. En cas de thrombose splanchnique, la polyglobulie peut être masquée par une carence martiale sur hémorragie digestive liée aux varices œsophagiennes, mais aussi par l’hypersplénisme lié à l’hypertension portale. En cas de carence martiale, une recharge prudente de la carence peut révéler la polyglobulie. En cas d’hypersplénisme, la mesure de la masse sanguine permettra le diagnostic en mettant en évidence l’augmentation du volume globulaire.
La myélofibrose primitive et en particulier la myélofibrose préfibrotique peuvent se présenter initialement comme une thrombocytémie essentielle. La clinique trouvera plus souvent des signes généraux et une splénomégalie. Sur le frottis, on pourra noter une érythromyélémie, des dacryocytes. La recherche des mutations n’aidera guère au diagnostic différentiel, puisqu’on les retrouve également dans cette pathologie. C’est la biopsie ostéomédullaire qui permettra de trancher définitivement avec la présence d’une hypercellularité, d’atypie des mégacaryocytes et éventuellement de fibrose réticulinique, voire collagénique de grade 2 ou 3 (ou de grade 1 dans la myélofibrose préfibrotique).
Syndromes myélodysplasiques
Il existe dans certains syndromes myélodysplasiques une thrombocytose associée. On peut citer le syndrome 5q-, le syndrome myélodysplasique/myéloprolifératif – avec sidéroblastes en couronne – et thrombocytose. Le myélogramme et la cytogénétique médullaire permettront le diagnostic.Thrombocytoses familiales
Elles restent exceptionnelles, résultat de la présence d’une mutation activatrice portant sur le gène de la thrombopoïétine ou de son récepteur (MPL).Complications et évolution
Thromboses artérielles et veineuses
Leur mécanisme n’est pas totalement élucidé : participent l’hyperplaquettose, mais aussi des anomalies intrinsèques des cellules mutées, hématologiques ou endothéliales, entraînant un état prothrombogène et inflammatoire.Au même titre que la polyglobulie de Vaquez, il faut considérer la thrombocytémie essentielle comme un facteur de risque cardiovasculaire à part entière, qui majore le risque d’athérosclérose et notamment d’accident coronarien, d’accident vasculaire cérébral.
Les thromboses veineuses pouvant compliquer la thrombocytémie essentielle touchent parfois des territoires rares : thrombose splanchniques (thrombose porte, syndrome de Budd-Chiari), thromboses veineuses cérébrales. Le risque thrombotique post-opératoire est également augmenté et prolongé, justifiant une prophylaxie renforcée.
Hémorragies
Le risque hémorragique est lié à la thrombopathie (syndromes de Willebrand acquis notamment) et augmente avec le taux de plaquettes (lié surtout aux thrombocytose extrêmes, > 1 500 G/L). L’antiagrégation plaquettaire doit être évitée en cas de thrombocytose >1 000 G/L, tant que le chiffre de plaquettes n’est pas réduit par un traitement cytoréducteur.Risque évolutif à long terme
L’évolution vers la myélofibrose ou la leucémie aiguë est possible après plusieurs dizaines d’années, mais ce risque est un peu moindre que dans les polyglobulies de Vaquez. Ces leucémies aiguës secondaires ont en revanche également un pronostic très sombre.Pronostic
Le pronostic est marqué principalement par les complications thrombo-hémorragiques mais aussi par l’évolution phénotypique.Le risque thrombotique est diminué par un bon contrôle de la pathologie par les traitements cytoréducteurs mais n’est pas ramené à celui de la population générale. Le risque hémorragique est augmenté par l’antiagrégation ou l’anticoagulation. Aucun traitement n’a fait preuve d’une efficacité sur le risque de transformation.
L’espérance de vie est quasiment identique à celle de la population générale.
Principe de traitement
Antiagrégation et anticoagulation
L’aspirine à dose antigrégante (100 mg x 1 ou x 2/j) peut être utilisée pour prévenir le risque thromboembolique dans la thrombocytémie essentielle, même si le niveau de preuve quant à son efficacité est moins formelle que pour la polyglobulie de Vaquez.Les anticoagulants n’ont leur place qu’en cas de survenue de thrombose veineuse. Le traitement est alors à envisager au long cours, sauf contre-indication.
Cytoréducteurs médicamenteux
Une cytoréduction médicamenteuse est indiquée chez les patients à haut risque thrombotique, défini par la présence d’un ou plusieurs des facteurs de risque suivants : âge supérieur à 60 ans, antécédent de thrombose, thrombocytose supérieure à 1 500 G/L. L’objectif est alors de normaliser le chiffre des plaquettes.Ils ont fait la preuve de leur efficacité pour réduire le risque thrombotique ou hémorragique, mais aucun n’a pour l’instant fait preuve d’une efficacité à réduire le risque de transformation en myélofibrose ou leucémie aiguë. Certains posent même le problème d’un potentiel leucémogène.
Un médicament a l’indication en 1re ligne, l’hydroxyurée.
Un médicament a l’indication en 2e ligne, l’anagrélide.
Le peginterféron alfa-2a est parfois également utilisé en 1re ligne (ou ultérieurement), selon les recommandations internationales des sociétés savantes, mais en dehors de toute autorisation de mise sur le marché.
Anagrélide : traitement de 2e ligne en cas de résistance ou d’intolérance à l’hydroxyurée. Il est parfois également proposé en alternative à l’hydroxyurée chez l’adulte jeune, du fait de son absence vraisemblable de risque leucémogène. Au 1er plan des effets secondaires : céphalées et vertiges, troubles digestifs, et tachycardie et palpitations. Un bilan préthérapeutique et un suivi cardiologique sont nécessaires du fait d’un risque d’événements indésirables cardiaques graves, en dehors des palpitations très fréquentes en début de traitement mais sans gravité.•
POINTS FORTS À RETENIR
La LMC, avant l’avènement des inhibiteurs de tyrosine kinase (ITK), avait une histoire naturelle très grave, avec une inexorable évolution vers la transformation en leucémie aiguë.
Le diagnostic de LMC repose sur une NFS caractéristique (hyperleucocytose à PNN, basophilie, myélémie ± thrombocytose) associée à l’existence d’un transcrit de fusion BCR-ABL1, reflet d'une translocation t(9;22).
Le pronostic de la LMC a été révolutionné par l’arrivée des ITK, c’est maintenant une maladie considérée comme une maladie chronique, et les patients ont une espérance de vie quasi identique à la population générale.
La PV et la TE sont des hémopathies myéloïdes clonales chroniques, donc le principal risque évolutif à court et moyen terme est le risque thrombotique veineux ou artériel.
Le diagnostic de PV repose avant tout sur des valeurs d’Hb et Ht augmentées et la présence d’une mutation de JAK2 (JAK2 V617F dans 95 % des cas, ou mutation de l’exon 12 de JAK2 dans 2% des cas). La majorité des polyglobulies secondaires peuvent être éliminées avec une échographie abdominale, un interrogatoire à orientation pneumologique et des gaz du sang.
Le diagnostic de thrombocytémie essentielle se fait après avoir tout d’abord éliminé une thrombocytose réactionnelle (carence martiale, syndrome inflammatoire). On retrouve dans 85 % des cas une mutation JAK2V617F ou une mutation de CALR ou de MPL. En l’absence de ces mutations, la biopsie ostéomédullaire permet le diagnostic des TE dites « triple négatives » mais également le diagnostic différentiel avec les autres SMP non LMC.
Le traitement des PV et des TE vise avant tout à réduire le risque thrombotique. Il associe, selon le niveau de risque, anti-agrégation plaquettaire (ou anticoagulation en cas de thrombose veineuse) ± cytoréduction. Un point important est la prise en charge en parallèle des autres FDRCV.
 Encadrés
Encadrés