Les syndromes parkinsoniens atypiques sont des syndromes parkinsoniens « impurs », où la triade classique (bradykinésie, rigidité, tremblement de repos) est associée à des signes ou symptômes d’alerte appelés « drapeaux rouges ». On appelle ainsi un symptôme inattendu ou atypique pour une maladie de Parkinson débutante. La présence de ces drapeaux rouges amène à reconsidérer le diagnostic de maladie de Parkinson idiopathique et dans certains cas à prescrire des examens complémentaires.
Quand penser à un syndrome parkinsonien atypique ?
– une progression rapide ;
– un début symétrique ;
– des troubles de l’équilibre, des troubles posturaux, la survenue précoce de chutes ;
– des signes précoces de dysautonomie (incontinence urinaire, troubles érectiles, hypotension orthostatique sévère), d’atteinte pseudobulbaire, ou d’atteinte cognitive ;
– un syndrome cérébelleux ;
– un syndrome pyramidal ;
– des troubles oculomoteurs ;
– des signes corticaux (apraxie, aphasie, troubles sensitifs) ;
– des hallucinations ;
– l’absence de réponse ou une réponse pauvre ou courte au traitement dopaminergique ;
– des anomalies en imagerie cérébrale (tomodensitométrie, imagerie par résonance magnétique [IRM]).
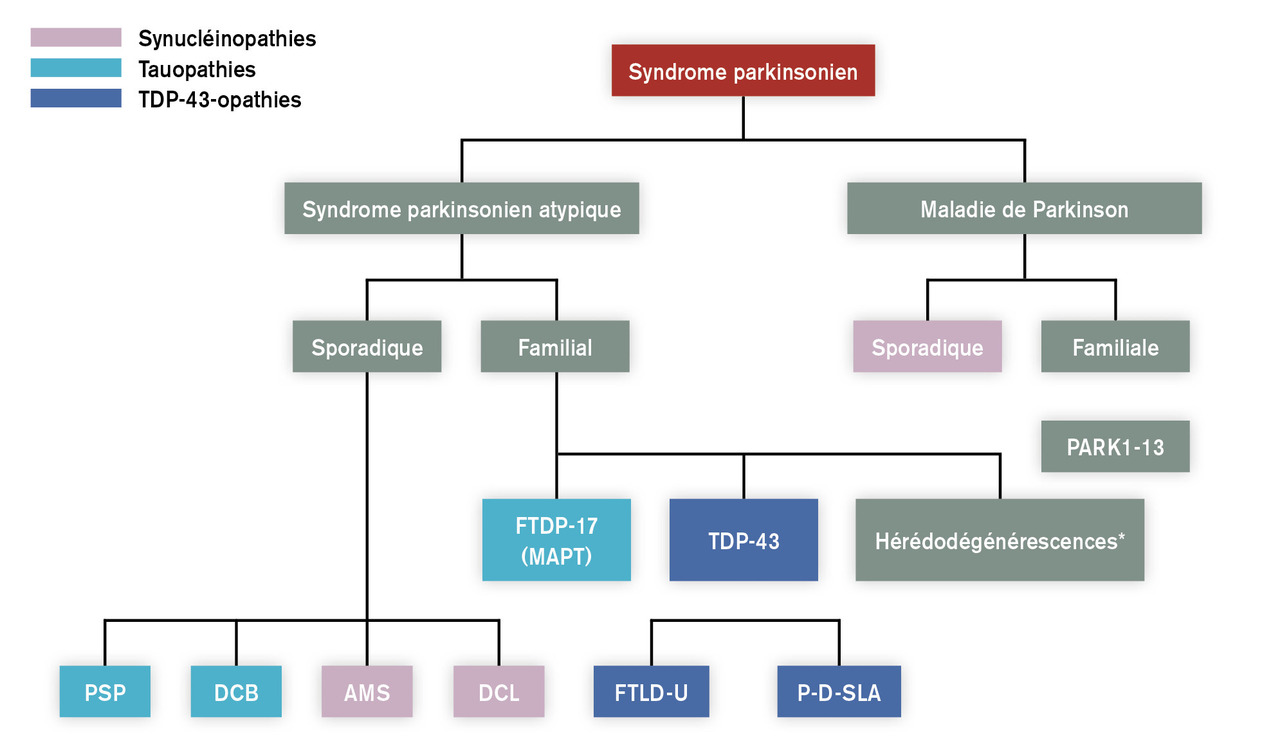
Trois catégories de syndromes parkinsoniens atypiques
– les syndromes parkinsoniens sporadiques dégénératifs ;
–les syndromes parkinsoniens familiaux (différentes formes de démence fronto-temporale avec parkinsonisme [FTDP-17 ou TDP-43-opathies], la maladie de Huntington, les ataxies spinocérébelleuses 2, 3, 17, la maladie de Wilson pour les plus connues) ;
– les syndromes parkinsoniens secondaires (post-neuroleptiques, vasculaires, post-traumatiques, par hydrocéphalie chronique, post-encéphalitiques, causes anoxiques, toxiques, métaboliques).
Syndromes parkinsoniens atypiques neurodégénératifs
L’atrophie multisystématisée et la démence à corps de Lewy sont, comme la maladie de Parkinson, des synucléinopathies, caractérisées par des agrégats intracellulaires insolubles d’alphasynucléine. Les taupathies ou tauopathies regroupent les affections au cours desquelles des agrégats intracellulaires de protéine tau sont observés, comme dans la paralysie supranucléaire progressive, la dégénérescence corticobasale et la démence frontotemporale avec parkinsonisme.
Pour ces quatre pathologies, des critères permettent d’évaluer le degré de fiabilité du diagnostic posé (
Atrophie multisystématisée2, 3
Il s’agit d’une synucléinopathie sporadique dont l’incidence est de 3 cas/100 000/an et le sex-ratio de 1,4 homme pour 1 femme. L’âge médian au diagnostic est de 60 ans (la maladie débute très rarement après 75 ans). La survie médiane varie de 6 à 10 ans selon les études.Comme son nom l’indique, plusieurs symptômes appartenant à plusieurs systèmes sont associés : parkinsonisme, signes autonomiques, syndrome cérébelleux, signes pyramidaux.
La dysautonomie est présente chez tous les patients ; il peut s’agir d’une hypotension orthostatique souvent sévère, de troubles urinaires (incontinence), d’une dysfonction érectile chez l’homme, de troubles digestifs (constipation), parfois d’un acrosyndrome. Les symptômes dysautonomiques apparaissent souvent les premiers, associés ou non à des symptômes moteurs.
Il existe deux formes d’atrophie multisystématisée (AMS) :
– prédominance du syndrome parkinsonien (AMS-P), la plus fréquente : dysautonomie et syndrome akinétorigide symétrique axial, progression plus rapide que pour la maladie de Parkinson ; instabilité posturale, chutes précoces (dans les 3 ans suivant le début de la maladie, renforcée par la présence du syndrome parkinsonien et d’une éventuelle hypotension orthostatique, mais les chutes sont moins précoces qu’au cours de la paralysie supranucléaire progressive) ; tremblement de repos rare ; myoclonies stimulo-induites ; faible réponse à la L dopa (environ 30 %) ; dyskinésies axiales, orofaciales parfois observées ;
– prédominance du syndrome cérébelleux (AMS-C) : ataxie à la marche, dysarthrie cérébelleuse, dysmétrie, troubles oculaires cérébelleux (anomalie des saccades, nystagmus) ; le syndrome parkinsonien est discret, voire absent, en début de maladie.
D’autres caractéristiques cliniques sont communes aux deux formes :
– anomalie de la posture : camptocormie, Pisa syndrome (déviation latérale du rachis), antérocolis ;
– myoclonies, tremblement myoclonique ;
– atteinte bulbaire : dysphagie, dysarthrie, dysphonie sévères ;
– signes pyramidaux : réflexes ostéotendineux vifs diffusés, signe de Babinski ;
– troubles du sommeil : stridor inspiratoire nocturne, apnées du sommeil, trouble du comportement en sommeil paradoxal (agitation nocturne au cours de cette phase du sommeil où l’on rêve et où normalement il existe une atonie musculaire, absente ici chez les patients qui, en conséquence, vivent leur rêve) ;
– rires et pleurs inappropriés ;
– progression rapide ;
– un syndrome frontal peut être présent, mais pas de démence ;
– faible réponse à la L-dopa.
Paralysie supranucléaire progressive, ou maladie de Steele-Richardson-Olszewski4, 5
C’est une tauopathie sporadique dont la prévalence est de 6,4 cas/100 000/an et le sex-ratio de 3 hommes pour 2 femmes. L’âge de début moyen est de 65 ans et la durée moyenne d’évolution de 7 ans.La forme classique associe une ophtalmoplégie supranucléaire verticale à une instabilité posturale importante, responsable de chutes dans la première année d’évolution des symptômes.
Les symptômes en sont :
– ophtalmoplégie supranucléaire, paralysie des mouvements oculaires dont la verticalité du regard est la plus atteinte, notamment le regard vers le bas. Impression de fixité du regard, yeux écarquillés et une lenteur des saccades oculaires ;
– instabilité posturale précoce avec chutes ;
– syndrome parkinsonien axial et symétrique, rigide surtout ;
– à la marche, on note un freezing précoce (impression que les pieds restent collés au sol), genoux en extension ;
– syndrome frontal pouvant évoluer vers une démence : atteinte sous-corticofrontale, ralentissement psychomoteur, troubles attentionnels et syndrome dysexécutif, grasping, persévération, désinhibition, signe de l’applaudissement ;
– apathie (manque de motivation) ;
– syndrome pseudobulbaire : dysarthrie, dysphagie, rires et pleurs spasmodiques ;
– dystonie : rétrocolis, blépharospasme, apraxie à l’ouverture des yeux ;
– faible réponse à la L-dopa.
Une paralysie supranucléaire progressive classique débutant après 63 ans avec une dysphagie précoce et des troubles cognitifs précoces a un pronostic négatif.
Dégénérescence corticobasale6
Les données épidémiologiques de cette tauopathie sporadique, maladie rare, sont peu nombreuses. Le sex-ratio est de 1/1, avec un âge de début médian de 63 ans.Elle se caractérise par un syndrome parkinsonien akinétorigide asymétrique non-dopa-sensible associé à une apraxie motrice et à des mouvements anormaux involontaires.
Le caractère asymétrique est un des éléments clés du diagnostic, qui peut parfois être un facteur confondant avec la maladie de Parkinson. La très faible dopa-sensibilité permet de l’en distinguer.
Le mode d’entrée dans la maladie est déterminé par la prédominance du volet moteur ou du volet cognitif :
– volet prédominant « basal » ou sous-cortical par atteinte des ganglions de la base : composante motrice au premier plan, généralement très asymétrique, voire unilatérale, qui peut comporter un tremblement de repos, une dystonie segmentaire ou hémicorporelle, une akinésie et rigidité extrapyramidale ;
– volet prédominant cortical par atteinte des régions pariétorolandiques : troubles apraxiques, phénomène de la main étrangère (« alien limb »), troubles sensitifs élaborés (astéréognosie, apallesthésie, agraphesthésie), myoclonies spontanées ou réflexes.
Des troubles cognitifs et comportementaux peuvent être associés : syndrome dysexécutif, troubles attentionnels, apraxie buccofaciale, aphasie non fluente progressive avec nette dissociation automatico-volontaire.
Démence à corps de Lewy7
L’anatomopathologie de cette synucléinopathie sporadique est complexe : coexistence de corps de Lewy avec lésions vasculaires ou lésions caractéristiques de la maladie d’Alzheimer. Sa prévalence est d’environ 0,7 % après 65 ans, avec un sex- ratio de 1,9 homme pour 1 femme. L’âge d’apparition de la maladie se situe entre 60 et 90 ans.C’est la deuxième cause de démence neurodégénérative, après la maladie d’Alzheimer. Elle représente 20% des maladies neurodégénératives et se caractérise, au cours de la première année, par l’association d’un syndrome démentiel fluctuant à prédominance frontale, d’hallucinations visuelles et d’un syndrome parkinsonien.
Les symptômes sont :
– des troubles cognitifs (60 % des patients) ;
– des fluctuations cognitives et attentionnelles ;
– un syndrome parkinsonien (25 % des patients) bilatéral, léger, symétrique fréquemment akinétorigide, avec une réponse partielle à la L-dopa ;
– des hallucinations visuelles, précoces, complexes, récurrentes, pouvant au début être bien critiquées ;
– une dysautonomie cardiovasculaire, digestive et urogénitale fréquente ;
– des troubles du comportement en sommeil paradoxal ;
– une hypersensibilité aux neuroleptiques (effets indésirables dangereux des neuroleptiques de 1re génération : sédation profonde, aggravation du syndrome parkinsonien, aggravation de l’altération cognitive, chutes).
Syndromes parkinsoniens familiaux
Syndromes parkinsoniens atypiques secondaires
Syndrome parkinsonien iatrogène
De nombreux médicaments peuvent être à l’origine d’un syndrome parkinsonien.8 Le tableau clinique habituellement rapporté est un syndrome akinétorigide plutôt axial ou symétrique associé à un tremblement d’attitude. La prise de neuroleptiques sous toutes leurs formes (antipsychotiques, antiémétiques, hypnotiques, contre les bouffées de chaleurs, etc.) doit systématiquement être recherchée. Les symptômes sont réversibles à l’arrêt du traitement.Syndrome parkinsonien lésionnel par atteinte des noyaux gris centraux
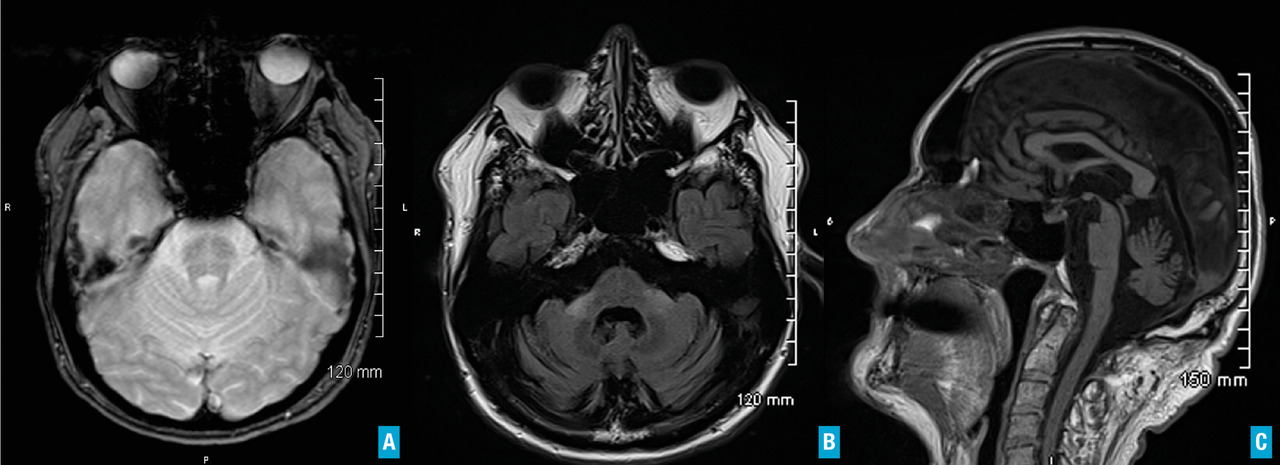
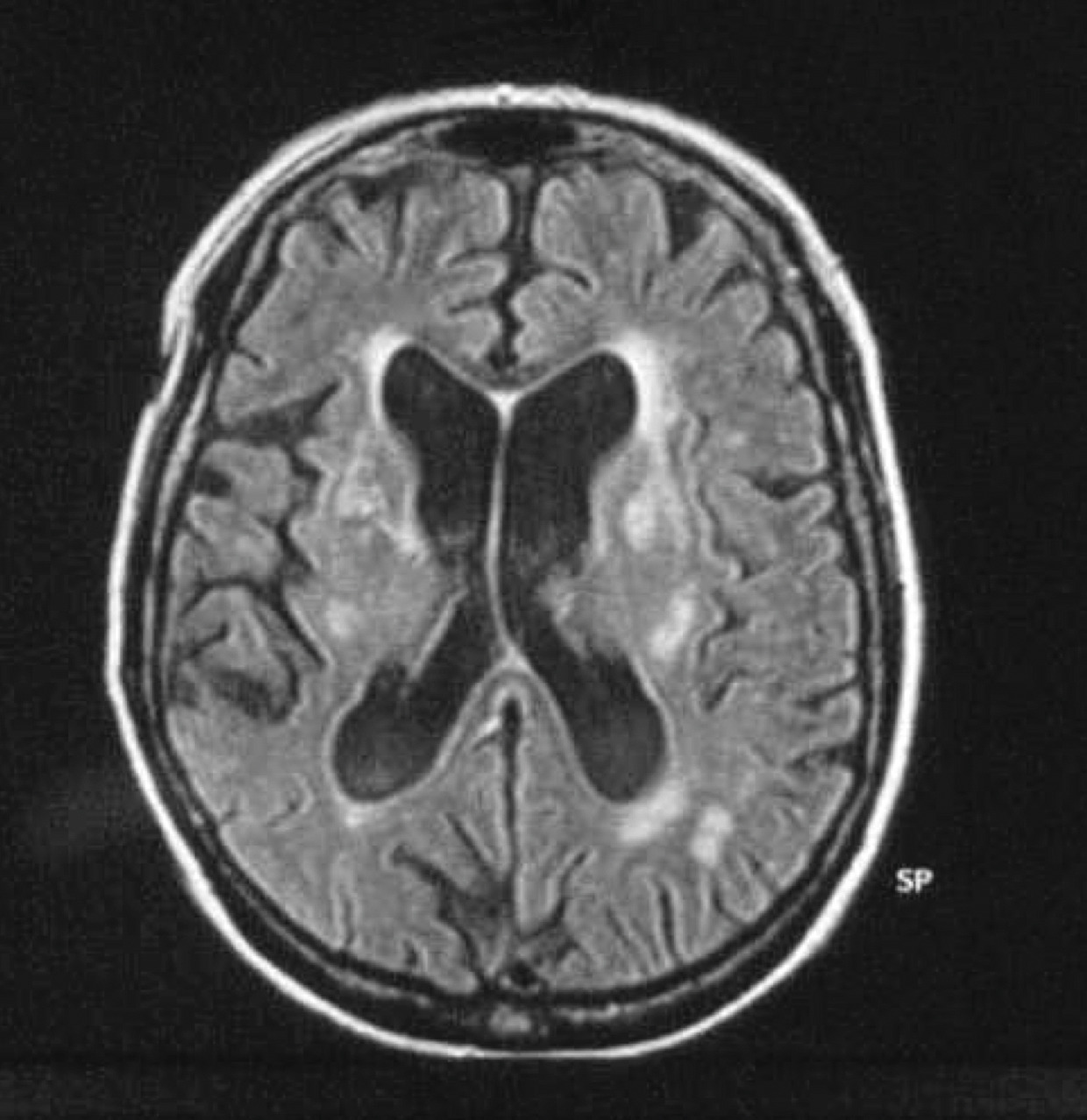
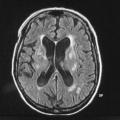
Un dysfonctionnement des noyaux gris centraux, qu’il soit d’origine vasculaire, infectieuse, tumorale ou associé à une hydrocéphalie, peut entraîner un syndrome parkinsonien.Syndrome parkinsonien d’origine vasculaire. Il est la conséquence de lésions vasculaires multiples affectant les noyaux gris centraux ou la substance blanche. L’IRM cérébrale permet de confirmer le diagnostic. Les caractéristiques cliniques sont : un syndrome parkinsonien plutôt symétrique, peu dopa-sensible ; prédominance des troubles aux membres inférieurs, marche avec freezing précoce ; syndrome pseudobulbaire.
Syndrome parkinsonien d’origine toxique. Les intoxications au monoxyde de carbone et au manganèse (exposition directe, accumulation intracérébrale chez un patient avec shunt portosystémique, abus de toxiques comme l’éphédrone) peuvent induire un syndrome parkinsonien. Les noyaux gris centraux sont très sensibles à l’anoxie et à certaines substances. L’anamnèse et l’imagerie permettent de confirmer le diagnostic.
Hydrocéphalie à pression normale. La triade classique de l’hydrocéphalie (troubles cognitifs, troubles de la marche avec rétropulsion, troubles urinaires) s’accompagne fréquemment d’un syndrome parkinsonien. L’imagerie et la faible réponse au traitement dopaminergique orientent le diagnostic.
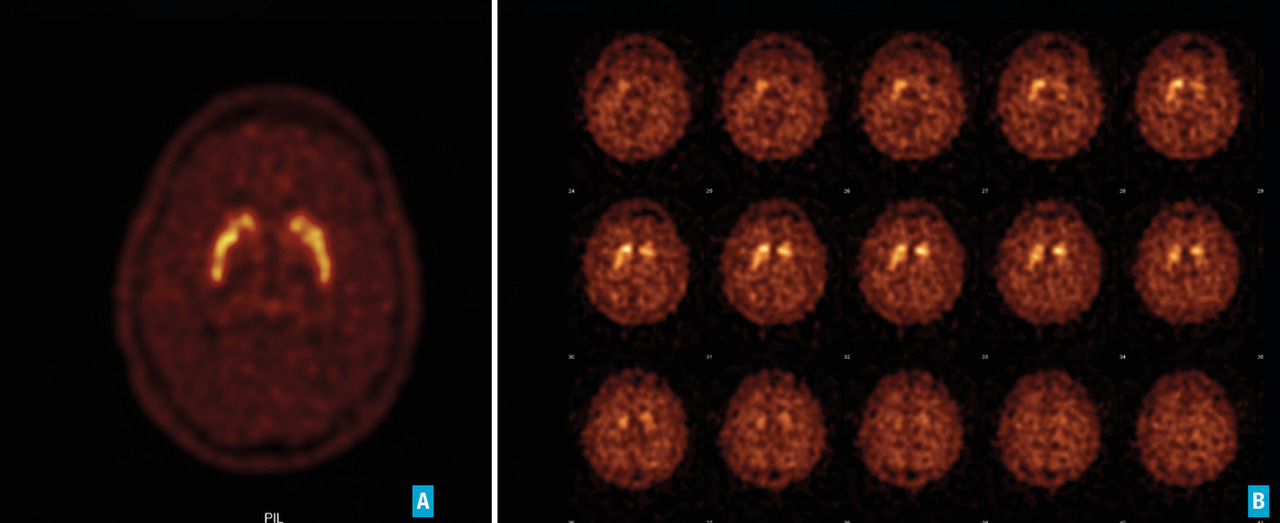
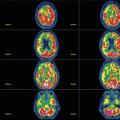
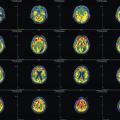
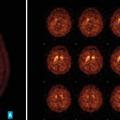
Quels examens complémentaires ?
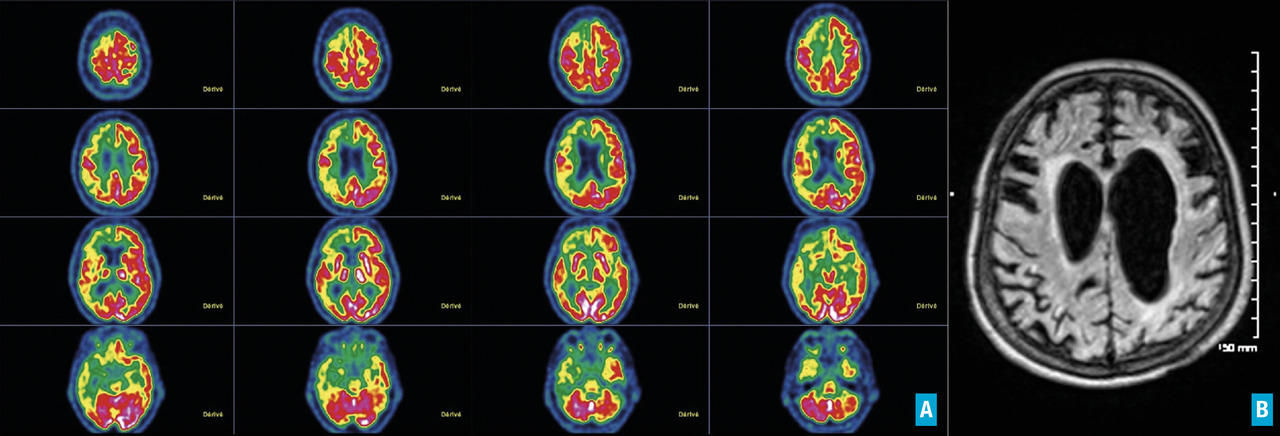
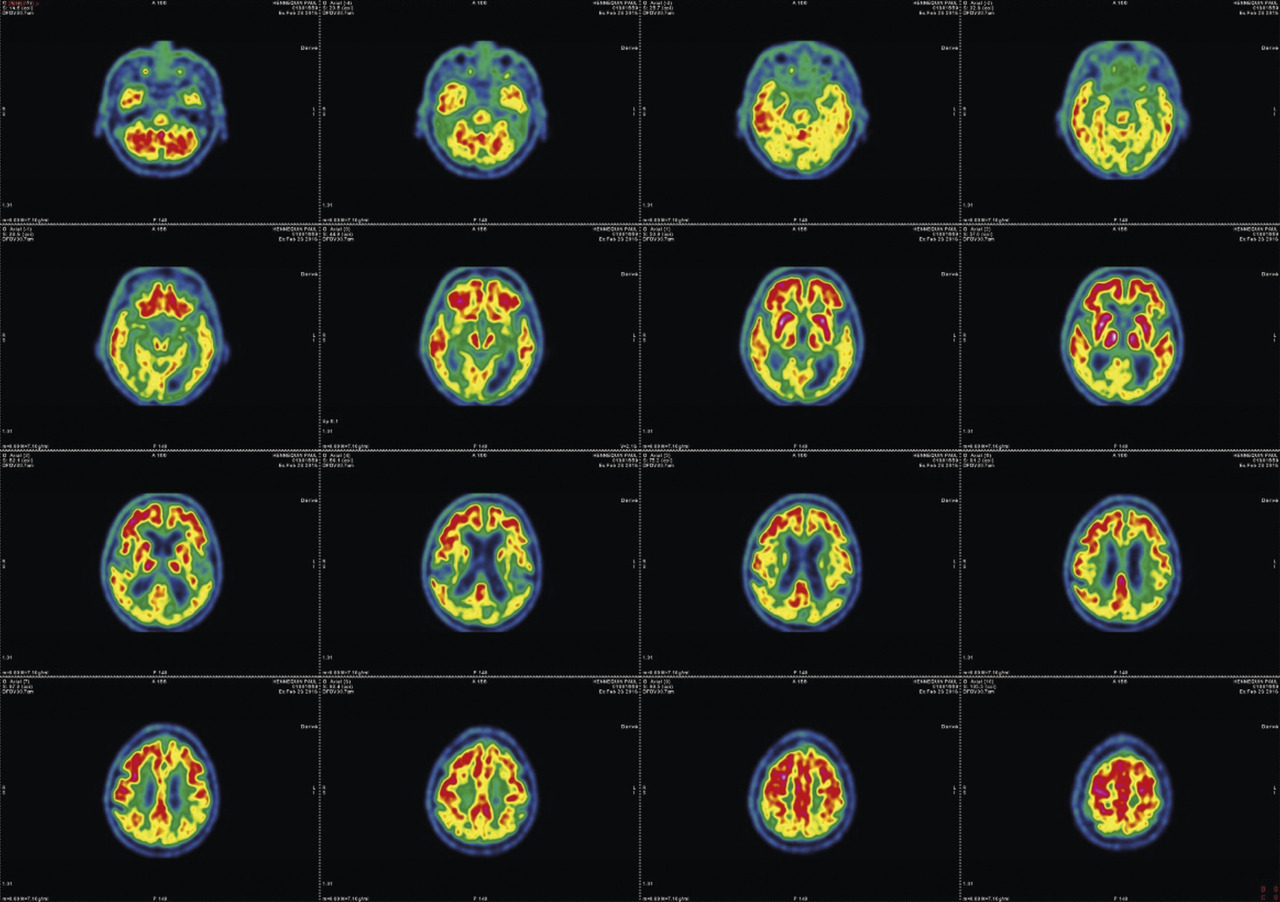
L’imagerie cérébrale morphologique et fonctionnelle (
Évolution et prise en charge
Certaines pathologies ont un traitement spécifique :
– maladie de Wilson : traitement chélateur du cuivre, D-pénicillamine, zinc, trientine ;
– hydrocéphalie à pression normale : traitement chirurgical par systèmes de dérivation du liquide cérébrospinal ;
– syndrome parkinsonien iatrogène : arrêt du médicament en cause).
Pour les autres, le traitement des syndromes parkinsoniens repose principalement sur une prise en charge symptomatique et multidisciplinaire, faisant intervenir médecin traitant, neurologue, ergothérapeute, kinésithérapeute, orthophoniste, psychologue, soins palliatifs, parfois cardiologue, urologue, gastroentérologue. La prise en charge sociale est essentielle : importance du soutien familial, prise en charge des aidants, hospitalisation de répit, anticipation d’une institutionnalisation. L’ensemble des structures de soins développés pour la maladie d’Alzheimer peut être envisagé pour les patients atteints de démence à corps de Lewy.
Traitement symptomatique : quelques exemples
Pour lutter contre l’hypotension orthostatique, les règles hygiénodiététiques (apport quotidien suffisant d’eau, régime salé, port de bas de contention, éviction des traitements hypotenseurs) peuvent être complétées, si l’hypotension persiste, par deux médicaments qui ont l’autorisation de mise sur le marché (AMM) dans cette indication : lamidodrine et la fludrocortisone.
En cas de troubles urinaires peuvent être prescrits un alphabloquant ou un anticholinergique (avec un passage le plus limité possible de la barrière hémato-encéphalique) ou une injection de toxine botulinique.
Contre la dystonie, le blépharospasme, l’apraxie à l’ouverture des yeux, des injections de toxine botulique peuvent aussi être proposées.
Contre les troubles posturaux et de la marche, la kinésithérapie est indispensable.
Dans les troubles cognitifs (maladie à corps de Lewy), les inhibiteurs de l’acétylcholinestérase peuvent apporter un bénéfice symptomatique supérieur à ce qui est observé dans la maladie d’Alzheimer. Ils améliorent modestement les fluctuations cognitives, les capacités attentionnelles, mnésiques, les troubles du comportement et parfois les fonctions exécutives. Seule la rivastigmine, dont le remboursement n’est plus assuré par la Sécurité sociale depuis 2018, possède l’AMM.
Pour limiter les troubles du comportement, il est préférable d’éviter les neuroleptiques, ou de favoriser les neuroleptiques avec le plus faible risque d’entraîner un syndrome parkinsonien (clozapine ou éventuellement quétiapine). En cas de prescription de clozapine, la surveillance de l’hémogramme est hebdomadaire à l’initiation, en raison du risque d’agranulocytose.
Pour traiter les hallucinations ou un syndrome pseudopsychotique invalidant, un antipsychotique atypique (clozapine, quétiapine) peut être prescrit.
En cas de trouble du comportement en sommeil paradoxal, la mélatonine ou une benzodiazépine (clonazépam) peut être donnée au coucher à dose minimale efficace.
En cas de dépression, il faut privilégier les inhibiteurs de la recapture de la sérotonine et éviter les tricycliques.
Perspectives
Concernant la paralysie supranucléaire progressive, une immunothérapie anti-tau est à l’étude.
Ce qu’il faut retenir
Le diagnostic des syndromes parkinsoniens atypiques reste difficile au stade précoce.
Le syndrome parkinsonien s’associe à d’autres symptômes : ces « drapeaux rouges » doivent être recherchés à l’interrogatoire et à l’examen physique.
Les progrès de l’imagerie fonctionnelle apportent une aide au diagnostic.
Contrairement à la maladie de Parkinson, les signes moteurs répondent peu ou pas au traitement dopaminergique.
Moins bon pronostic : la progression des signes est plus rapide et la durée de survie est diminuée par rapport à la maladie de Parkinson.
Les causes curables doivent être identifiées : prise de neuroleptique, hydrocéphalie, maladie de Wilson.
La certitude diagnostique, obtenue par l’anatomopathologie, n’est pas disponible du vivant du patient.
La prise en charge est multidisciplinaire. Pas de traitement curatif disponible.
Quelques liens utiles
Centre de référence de l’atrophie multisystématisée : CHU Toulouse et Bordeaux.
https://www.chu-toulouse.fr/-centre-de-reference-de-l-atrophie-multisystematisee-
Association de malades atteints d’atrophie multisystématisée
Association de soutien à la paralysie supranucléaire progressive et dégénérescence corticobasale : association PSP France
2. Meissner WG, Fernagut P, Dehay B, Péran P, Pavy-Le Traon A, et al. Multiple system atrophy: recent developments and future perspectives. Mov Disord 2019;34:1629-42.
3. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670-6.
4. Armstrong MJ. Progressive supranuclear palsy: an update. Curr Neurol Neurosci Rep 2018;18:12.
5. Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria: MDS clinical diagnostic criteria for PSP. Mov Disord 2017;32:853-64.
6. Armstrong MJ, Litvan I, Lang AE, Bak TH, Bathia KP, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496-503.
7. McKeith IG, Dickson DW, Lowe J, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 2005;65:1863-72.
8. Bondon-Guitton E, Perez-Lloret S, Bagheri H, Brefel C, Rascol O, Montastruc JL. Drug-induced parkinsonism: a review of 17 years’ experience in a regional pharmacovigilance center in France. Mov Disord 2011;26:2226-31.
 Encadrés
Encadrés