Les toxidermies sont des réactions d’hypersensibilité retardée aux médicaments. Pour un principe actif donné, 0,1 à 1 % des patients feront une toxidermie, parmi lesquels 0,1 à 0,3 % une forme grave à hospitaliser. Savoir les repérer sans retard est crucial.
Toxidermies graves
Elles peuvent mettre en jeu le pronostic vital, soit par défaillance cutanée majeure (nécrose épidermique), soit via des complications viscérales. Leur suspicion impose une évaluation hospitalière pour juger de la nécessité ou non d’une hospitalisation. Ce sont la pustulose exanthématique aiguë généralisée (PEAG),1 le DRESS (Drug Reaction with Eosinophilia and Systemic Symptoms),2 les syndromes de Stevens-Johnson et de Lyell (spectre de la nécrolyse épidermique)3-5 et l’érythème pigmenté fixe (EPF) bulleux généralisé.6
La PEAG survient rapidement après l’introduction du médicament (1 à 11 jours).1 Elle associe fièvre élevée et exanthème du tronc et de la racine des membres se renforçant dans les plis. De larges plages érythémateuses sont surmontées de petites pustules millimétriques (lésions liquidiennes à contenu trouble) non folliculaires qui, en confluant, entraînent un décollement cutané superficiel (« pseudo-Nikolsky »).
À un stade tardif, seule une desquamation post-pustuleuse peut être observée.
Le bilan biologique retrouve une hyperleucocytose à polynucléaires neutrophiles. Certaines anomalies traduisent une atteinte viscérale : cytolyse hépatique, insuffisance rénale. L’atteinte pulmonaire est rare mais doit être recherchée. La mortalité est d’environ 1 %.
Les médicaments fréquemment imputés sont les antibiotiques (pénicillines, macrolides, pristinamycine), les inhibiteurs calciques (diltiazem), la terbinafine.
Prise en charge : application de dermocorticoïdes (DC) très forts et hospitalisation de courte durée parfois nécessaire pour une surveillance clinico-biologique rapprochée.
Le DRESS survient dans les 2 à 8 semaines après l’introduction du traitement suspect. Le patient est fébrile, et son état général altéré, un syndrome pseudoviral peut précéder l’éruption cutanée. L’exanthème couvre volontiers 90 % de la surface corporelle et s’associe à un œdème des extrémités (visage, mains, pieds) et à une polyadénopathie. Une hyperéosinophilie supérieure à 500/mm3 (souvent > 1 000/mm3) et/ou un syndrome mononucléosique sont évocateurs.
L’atteinte viscérale est fréquente : cytolyse, voire insuffisance hépatique, insuffisance rénale et, plus rarement, cytopénies au sein d’un syndrome d’activation macrophagique, myocardite ou péricardite, insuffisance respiratoire. Les réplications virales HHV6, EBV et/ou CMV sont fréquentes. Un antiviral peut être discuté dans de rares cas de résistance aux traitements usuels. La mortalité varie de 1 à 10 %.
Les médicaments fréquemment responsables sont l’allopurinol, les anticonvulsivants carbamazépine, phénobarbital, lamotrigine, les sulfamides antibactériens et la minocycline. L’hospitalisation est le plus souvent nécessaire pour surveiller les lésions viscérales.
Traitement : DC très fort (propionate de clobétasol 0,05 % : 30 g, soit 3 tubes, en 1 application par jour sur tout le corps pendant 10 jours suivi d’une décroissance progressive) ou corticothérapie générale (0,5 à 1 mg/kg/j puis baisse progressive) en cas d’atteinte viscérale grave.
Les syndromes de Stevens-Johnson et de Lyell (ou nécrolyse épidermique toxique ; NET)3-5 sont les toxidermies les plus létales, le taux de mortalité allant de 10 à 40 %. Ils surviennent dans les 4 à 28 jours suivant la prise médicamenteuse.
Ces syndromes se manifestent par une atteinte muqueuse pluri-orificielle (oculaire, buccale, ORL, génitale et anale), un exanthème maculopapuleux [EMP] violacé, douloureux, avec macules purpuriques, pseudo-cocardes évoluant vers le décollement cutané, et un signe de Nikolsky positif (décollement de la peau érythémateuse à la pression).
L’atteinte muqueuse précède généralement les lésions cutanées. Des bulles tendues au début évoluent vers de larges décollements en linge mouillé. Les patients sont altérés et fébriles.
Les médicaments les plus fréquemment incriminés sont les anticonvulsivants comme pour le DRESS, les sulfamides antibactériens (cotrimoxazole), les AINS (oxicams), la névirapine. Les complications immédiates sont dues à la défaillance cutanée (sepsis à point de départ cutané, troubles hydroélectrolytiques, de la thermorégulation) et aux conséquences de lésions respiratoires potentielles (atteinte bronchique pouvant aller jusqu’au syndrome de détresse respiratoire aiguë).
Les autres pathologies viscérales spécifiques (rein, foie) ou hématologiques (syndrome d’activation macrophagique) sont rares. Le transfert vers un centre hospitalier spécialisé est urgent.
Une prise en charge en réanimation est souvent nécessaire. À ce jour, aucun traitement immunomodulateur spécifique n’a réellement fait la preuve de son efficacité hormis les soins de support. Contre la douleur : morphiniques intraveineux, notamment avant les soins cutanés.
Après la phase aiguë, les séquelles sont nombreuses : cutanées (dyschromies, paresthésies), phanériennes (dystrophie unguéale, alopécie), ophtalmologiques (kératites, trichiasis – inversion des cils frottant sur la surface oculaire – allant jusqu’à la cécité), muqueuses autres (stomatologiques, génitales, pulmonaires…) et psychologiques (syndrome de stress post-traumatique).
L’EPF bulleux généralisé est une toxidermie rare et potentiellement grave, survenant principalement chez les patients âgés, souvent après prise répétée du médicament en cause (paracétamol, AINS, antibiothérapie).
Des lésions érythémateuses en patchs de différente taille peuvent confluer pour former un exanthème plus diffus. Un décollement cutané bulleux peut alors être observé, et le signe de Nikolsky est parfois positif.
L’atteinte muqueuse est possible, principalement buccale ou génitale. Les patients sont souvent altérés et fébriles. Les complications sont celles du terrain fragile et de la défaillance cutanée.6
La prise en charge requiert fréquemment une hospitalisation. Comme dans le syndrome de Lyell, pas de traitement spécifique hormis les soins de support.
La PEAG survient rapidement après l’introduction du médicament (1 à 11 jours).1 Elle associe fièvre élevée et exanthème du tronc et de la racine des membres se renforçant dans les plis. De larges plages érythémateuses sont surmontées de petites pustules millimétriques (lésions liquidiennes à contenu trouble) non folliculaires qui, en confluant, entraînent un décollement cutané superficiel (« pseudo-Nikolsky »).
À un stade tardif, seule une desquamation post-pustuleuse peut être observée.
Le bilan biologique retrouve une hyperleucocytose à polynucléaires neutrophiles. Certaines anomalies traduisent une atteinte viscérale : cytolyse hépatique, insuffisance rénale. L’atteinte pulmonaire est rare mais doit être recherchée. La mortalité est d’environ 1 %.
Les médicaments fréquemment imputés sont les antibiotiques (pénicillines, macrolides, pristinamycine), les inhibiteurs calciques (diltiazem), la terbinafine.
Prise en charge : application de dermocorticoïdes (DC) très forts et hospitalisation de courte durée parfois nécessaire pour une surveillance clinico-biologique rapprochée.
Le DRESS survient dans les 2 à 8 semaines après l’introduction du traitement suspect. Le patient est fébrile, et son état général altéré, un syndrome pseudoviral peut précéder l’éruption cutanée. L’exanthème couvre volontiers 90 % de la surface corporelle et s’associe à un œdème des extrémités (visage, mains, pieds) et à une polyadénopathie. Une hyperéosinophilie supérieure à 500/mm3 (souvent > 1 000/mm3) et/ou un syndrome mononucléosique sont évocateurs.
L’atteinte viscérale est fréquente : cytolyse, voire insuffisance hépatique, insuffisance rénale et, plus rarement, cytopénies au sein d’un syndrome d’activation macrophagique, myocardite ou péricardite, insuffisance respiratoire. Les réplications virales HHV6, EBV et/ou CMV sont fréquentes. Un antiviral peut être discuté dans de rares cas de résistance aux traitements usuels. La mortalité varie de 1 à 10 %.
Les médicaments fréquemment responsables sont l’allopurinol, les anticonvulsivants carbamazépine, phénobarbital, lamotrigine, les sulfamides antibactériens et la minocycline. L’hospitalisation est le plus souvent nécessaire pour surveiller les lésions viscérales.
Traitement : DC très fort (propionate de clobétasol 0,05 % : 30 g, soit 3 tubes, en 1 application par jour sur tout le corps pendant 10 jours suivi d’une décroissance progressive) ou corticothérapie générale (0,5 à 1 mg/kg/j puis baisse progressive) en cas d’atteinte viscérale grave.
Les syndromes de Stevens-Johnson et de Lyell (ou nécrolyse épidermique toxique ; NET)3-5 sont les toxidermies les plus létales, le taux de mortalité allant de 10 à 40 %. Ils surviennent dans les 4 à 28 jours suivant la prise médicamenteuse.
Ces syndromes se manifestent par une atteinte muqueuse pluri-orificielle (oculaire, buccale, ORL, génitale et anale), un exanthème maculopapuleux [EMP] violacé, douloureux, avec macules purpuriques, pseudo-cocardes évoluant vers le décollement cutané, et un signe de Nikolsky positif (décollement de la peau érythémateuse à la pression).
L’atteinte muqueuse précède généralement les lésions cutanées. Des bulles tendues au début évoluent vers de larges décollements en linge mouillé. Les patients sont altérés et fébriles.
Les médicaments les plus fréquemment incriminés sont les anticonvulsivants comme pour le DRESS, les sulfamides antibactériens (cotrimoxazole), les AINS (oxicams), la névirapine. Les complications immédiates sont dues à la défaillance cutanée (sepsis à point de départ cutané, troubles hydroélectrolytiques, de la thermorégulation) et aux conséquences de lésions respiratoires potentielles (atteinte bronchique pouvant aller jusqu’au syndrome de détresse respiratoire aiguë).
Les autres pathologies viscérales spécifiques (rein, foie) ou hématologiques (syndrome d’activation macrophagique) sont rares. Le transfert vers un centre hospitalier spécialisé est urgent.
Une prise en charge en réanimation est souvent nécessaire. À ce jour, aucun traitement immunomodulateur spécifique n’a réellement fait la preuve de son efficacité hormis les soins de support. Contre la douleur : morphiniques intraveineux, notamment avant les soins cutanés.
Après la phase aiguë, les séquelles sont nombreuses : cutanées (dyschromies, paresthésies), phanériennes (dystrophie unguéale, alopécie), ophtalmologiques (kératites, trichiasis – inversion des cils frottant sur la surface oculaire – allant jusqu’à la cécité), muqueuses autres (stomatologiques, génitales, pulmonaires…) et psychologiques (syndrome de stress post-traumatique).
L’EPF bulleux généralisé est une toxidermie rare et potentiellement grave, survenant principalement chez les patients âgés, souvent après prise répétée du médicament en cause (paracétamol, AINS, antibiothérapie).
Des lésions érythémateuses en patchs de différente taille peuvent confluer pour former un exanthème plus diffus. Un décollement cutané bulleux peut alors être observé, et le signe de Nikolsky est parfois positif.
L’atteinte muqueuse est possible, principalement buccale ou génitale. Les patients sont souvent altérés et fébriles. Les complications sont celles du terrain fragile et de la défaillance cutanée.6
La prise en charge requiert fréquemment une hospitalisation. Comme dans le syndrome de Lyell, pas de traitement spécifique hormis les soins de support.
Toxidermie bénigne ?
L’exanthème maculopapuleux (EMP) est souvent le premier motif de consultation, son incidence exacte n’est pas connue. C’est la principale expression cutanée de la toxidermie bénigne, mais il peut également être la manifestation initiale d’une forme grave.
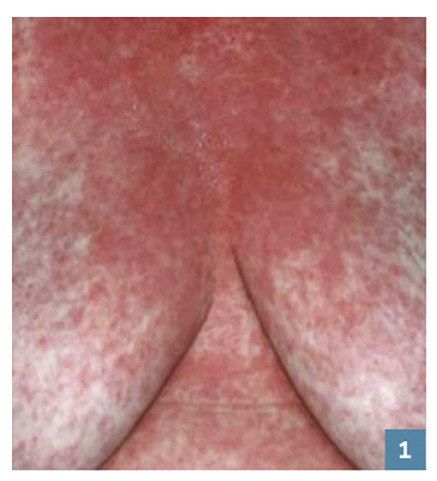
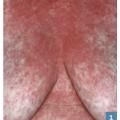
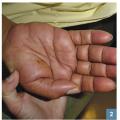
C’est une éruption érythémateuse diffuse faite de petites lésions maculopapuleuses s’étendant en quelques jours et pouvant confluer en grandes plages érythémateuses avec plus ou moins d’espaces de peau saine (fig. 1 ). Il survient de 7 à 21 jours après la prise, mais le délai est raccourci (24-48 h) si le patient est déjà sensibilisé. Son évolution est en général favorable, en 1 à 4 semaines après arrêt et élimination du principe actif, laissant la place à une desquamation sans séquelle.
Les signes orientant vers une toxidermie grave peuvent être discrets à ce stade (tableau ) : ils doivent être scrupuleusement recherchés. S’ils sont retrouvés ou en cas de doute, le patient doit être adressé vers un centre hospitalier en urgence.
L’analyse de l’exanthème peut orienter le diagnostic. Il est en grande nappe ou prédomine dans les plis dans la PEAG ; diffus et œdémateux aux extrémités dans le DRESS ; violacé et douloureux dans le syndrome de Lyell ; en grand patchs confluents dans l’EPF bulleux généralisé.
La fièvre élevée (> 38,5 °C) prolongée est le premier symptôme devant alerter. Elle est constatée dans toutes les toxidermies graves. Un pic fébrile lors d’un EMP bénin est possible, et d’autres arguments doivent orienter le clinicien.
La polyadénopathie fait suspecter un DRESS. Elle ne suffit pas à elle seule à poser le diagnostic et doit être associée à d’autres éléments cliniques.
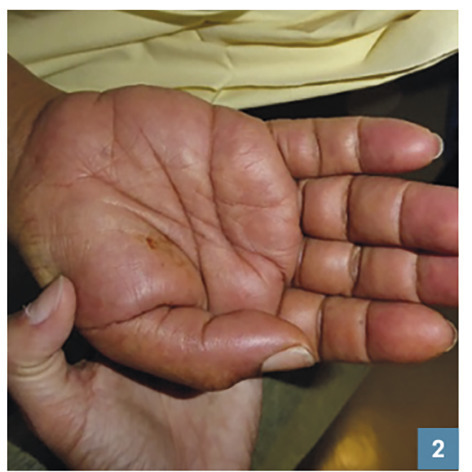
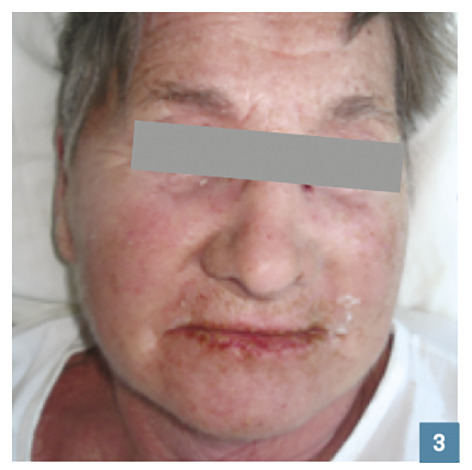
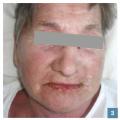
L’œdème érythémateux (fig. 2 et 3 ) du visage ou des mains doit faire craindre un DRESS. L’œdème des pieds, s’il est isolé, n’oriente pas obligatoirement vers un DRESS au vu des nombreuses causes possibles (insuffisance veineuse, cardiaque…).
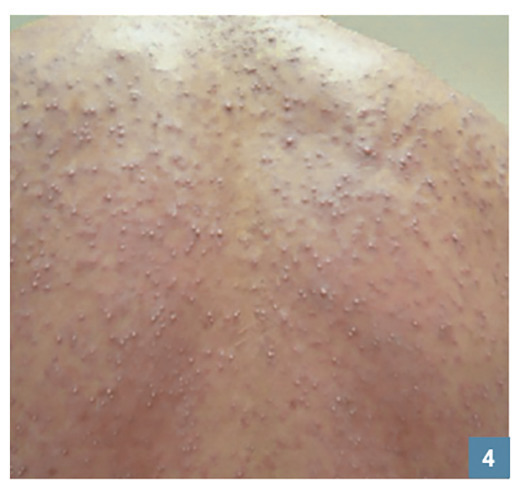
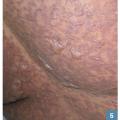
Des pustules éparses non folliculaires (fig. 4 ), dans les plis ou plus diffuses, font redouter une PEAG. Elles peuvent être confluentes en nappe et entraîner un décollement cutané superficiel. Elles sont très rarement retrouvées dans les DRESS.
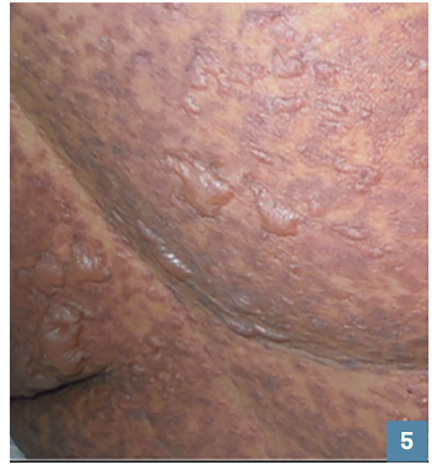
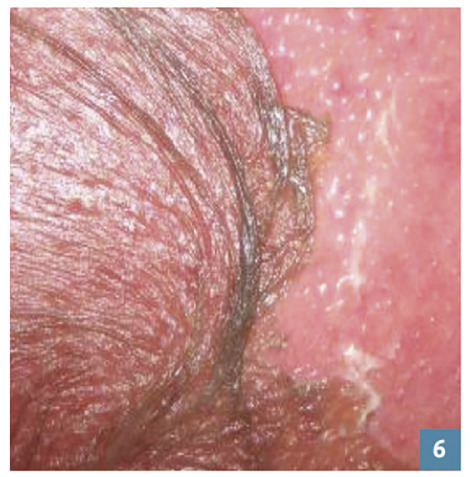
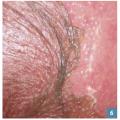
Les bulles cutanées (fig. 5 ) et le signe de Nikolsky (fig. 6 ) font suspecter une NET ou un EPF bulleux généralisé.
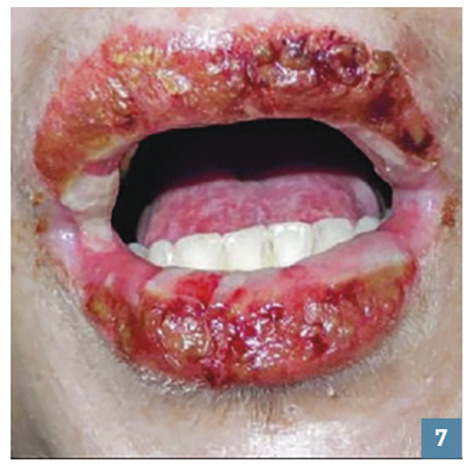
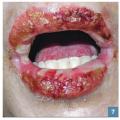
Une atteinte muqueuse (fig. 7 ) pouvant se manifester par des érosions ou bulles buccales, nasales, génitales ou anales ou par une conjonctivite doit également faire évoquer une NET débutante.
Un bilan biologique doit être fait autant que possible (tableau ).
Certaines anomalies motivent l’hospitalisation :
– une hyperéosinophilie> 1 000/mm3 ;
– un syndrome mononucléosique ;
– une cytolyse hépatique supérieure à 3 fois la norme ;
– une insuffisance rénale aiguë avec créatinine supérieure à 1,5 fois la norme ;
– des cytopénies.
Les toxidermies graves sont rares, mais leur aspect initial, parfois fruste, fait diagnostiquer par erreur un EMP bénin. Elles doivent être systématiquement recherchées. En cas de suspicion de toxidermie grave, le médicament imputable doit être interrompu, et le patient adressé en urgence vers un centre hospitalier spécialisé. Le cas doit être déclaré en pharmacovigilance.
La prévention de la récidive passe par une information claire du patient, de son entourage et de son MG. Une carte d’allergie précisant son type et les médicaments contre-indiqués doit être donnée à l’individu concerné.
Un bilan allergologique est systématiquement proposé, comprenant des tests épicutanés (patch tests) aux molécules imputées et à ceux de la même famille.
C’est une éruption érythémateuse diffuse faite de petites lésions maculopapuleuses s’étendant en quelques jours et pouvant confluer en grandes plages érythémateuses avec plus ou moins d’espaces de peau saine (
Les signes orientant vers une toxidermie grave peuvent être discrets à ce stade (
L’analyse de l’exanthème peut orienter le diagnostic. Il est en grande nappe ou prédomine dans les plis dans la PEAG ; diffus et œdémateux aux extrémités dans le DRESS ; violacé et douloureux dans le syndrome de Lyell ; en grand patchs confluents dans l’EPF bulleux généralisé.
La fièvre élevée (> 38,5 °C) prolongée est le premier symptôme devant alerter. Elle est constatée dans toutes les toxidermies graves. Un pic fébrile lors d’un EMP bénin est possible, et d’autres arguments doivent orienter le clinicien.
La polyadénopathie fait suspecter un DRESS. Elle ne suffit pas à elle seule à poser le diagnostic et doit être associée à d’autres éléments cliniques.
L’œdème érythémateux (
Des pustules éparses non folliculaires (
Les bulles cutanées (
Une atteinte muqueuse (
Un bilan biologique doit être fait autant que possible (
Certaines anomalies motivent l’hospitalisation :
– une hyperéosinophilie> 1 000/mm3 ;
– un syndrome mononucléosique ;
– une cytolyse hépatique supérieure à 3 fois la norme ;
– une insuffisance rénale aiguë avec créatinine supérieure à 1,5 fois la norme ;
– des cytopénies.
Les toxidermies graves sont rares, mais leur aspect initial, parfois fruste, fait diagnostiquer par erreur un EMP bénin. Elles doivent être systématiquement recherchées. En cas de suspicion de toxidermie grave, le médicament imputable doit être interrompu, et le patient adressé en urgence vers un centre hospitalier spécialisé. Le cas doit être déclaré en pharmacovigilance.
La prévention de la récidive passe par une information claire du patient, de son entourage et de son MG. Une carte d’allergie précisant son type et les médicaments contre-indiqués doit être donnée à l’individu concerné.
Un bilan allergologique est systématiquement proposé, comprenant des tests épicutanés (patch tests) aux molécules imputées et à ceux de la même famille.
Références
1. Sidoroff A, Halevy S, Bavinck JN, Vaillant L, Roujeau JC. Acute generalized exanthematous pustulosis (AGEP) - A clinical reaction pattern. J Cutan Pathol 2001;28:113‑9.
2. Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol 2013;169:1071‑80.
3. Duong TA, Valeyrie-Allanore L, Wolkenstein P, Chosidow O. Severe cutaneous adverse reactions to drugs. Lancet 2017;390:1996-2011.
4. Auquier-Dunant A, Mockenhaupt M, Naldi L, et al. Correlations between clinical patterns and causes of erythema multiforme majus, Stevens-Johnson syndrome, and toxic epidermal necrolysis: results of an international prospective study. Arch Dermatol 2002;138:1019-24.
5. HAS. Cente de référence des dermatoses bulleuses toxiques et toxidermies graves. Octobre 2017. https://bit.ly/2X7xS1t
6. Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol 2013;168:726‑32.
2. Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol 2013;169:1071‑80.
3. Duong TA, Valeyrie-Allanore L, Wolkenstein P, Chosidow O. Severe cutaneous adverse reactions to drugs. Lancet 2017;390:1996-2011.
4. Auquier-Dunant A, Mockenhaupt M, Naldi L, et al. Correlations between clinical patterns and causes of erythema multiforme majus, Stevens-Johnson syndrome, and toxic epidermal necrolysis: results of an international prospective study. Arch Dermatol 2002;138:1019-24.
5. HAS. Cente de référence des dermatoses bulleuses toxiques et toxidermies graves. Octobre 2017. https://bit.ly/2X7xS1t
6. Lipowicz S, Sekula P, Ingen-Housz-Oro S, et al. Prognosis of generalized bullous fixed drug eruption: comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol 2013;168:726‑32.
Dans cet article


essentiel
Signes généraux : fièvre, altération de l’état général, polyadénopathie.
Signes cliniques : œdème de la face ou des extrémités, infiltration cutanée, pustules, bulles ou signe de Nikolsky, atteinte muqueuse.
Signes biologiques : hyperéosinophilie, cytolyse hépatique, insuffisance rénale aiguë, cytopénie.