Introduction
Le don de sang, bénévole, anonyme et gratuit, fait suite à un entretien médical pré-don. C’est la première étape de la chaîne transfusionnelle. Deux types de don existent : le don de sang total et le don par aphérèse, permettant de séparer et sélectionner un composant du sang. Chaque don bénéficie d’une qualification biologique qui consiste en une série d’analyses biologiques, permettant de déterminer les caractéristiques immuno-hématologiques du produit mais aussi de dépister des maladies transmissibles par le sang.
L’étape de préparation permet ensuite de transformer un don en produit sanguin injectable suivant les caractéristiques des produits définies dans l’arrêté du 1er avril 2019.[1] L’ensemble de ces différentes étapes sont soumises au respect des bonnes pratiques transfusionnelles (BPT).[2]
Types de produits sanguins labiles et leurs indications (tableaux 1 et 2)
- soit à partir des couches leuco-plaquettaires issues d’un don de sang total de maximum 12 donneurs – on parle de mélange de concentrés de plaquettes ;
- soit à la suite d’un don par aphérèse issu d’un seul donneur ; on parle de concentrés de plaquettes d’aphérèse.
- la sécurisation par quarantaine, où le donneur est reconvoqué 60 jours après le don. De nouvelles analyses biologiques sont réalisées et, si les résultats sont négatifs, le produit peut être libéré ;
- l’illumination aux UVA en présence d’amotosalène (IA). L’amotosalène est un agent intercalant de l’ADN et de l’ARN qui, après illumination aux UVA, bloque la réplication des agents pathogènes bactériens et viraux.
Il existe également un plasma traité par solvant détergent faisant appel à un processus industriel et entrant dans la catégorie médicament. Il est géré par les pharmacies des établissements de soins et répond aux règles de la pharmacovigilance. En l’état actuel des connaissances, ces quatre plasmas sont considérés comme équivalents et ont les mêmes indications.
L’indication de la transfusion suit les recommandations de la Haute Autorité de santé (HAS), qui précisent les indications des produits sanguins labiles et les seuils transfusionnels.[3-5]
De plus, elle s’intègre aujourd’hui dans le « patient blood management » visant par une approche multidisciplinaire à optimiser les soins des patients susceptibles d’être transfusés, dans le but de maîtriser les besoins transfusionnels, les coûts de santé tout en augmentant la disponibilité des produits sanguins labiles.
Indications de la transfusion de concentrés de globules rouges
Cas particulier de la drépanocytose
Anciennement appelée anémie falciforme, la drépanocytose est la plus fréquente des maladies génétiques en France. Elle se caractérise par la présence d’une hémoglobine anormale, l’HbS (S pour sickle : faucille), entraînant des anomalies du globule rouge à l’origine d’une anémie chronique et de crises vaso-occlusives. Une transfusion simple sera indiquée dans les cas d’aggravation et d’intolérance clinique de l’anémie par rapport au taux basal de l’individu. Des échanges transfusionnels peuvent être indiqués en curatif ou en préventif pour les cas les plus sévères (antécédent d’accident vasculaire cérébral) afin de maintenir un taux d’HbS à un seuil infraclinique. La transfusion peut être parfois contre-indiquée, notamment en cas d’hémolyse post-transfusionnelle retardée, une complication redoutée et potentiellement mortelle. Certaines formes se caractérisent par une hémolyse intravasculaire massive, en raison de la destruction des globules transfusés et autologues, apparaissant entre 5 et 15 jours après la transfusion. Si cette complication est mal diagnostiquée, une transfusion supplémentaire peut exacerber l’hémolyse et les symptômes cliniques, et mettre en jeu le pronostic vital.[6]
Indications de la transfusion de plaquettes
Les seuils de numération plaquettaire justifiant la transfusion dans un contexte périopératoire sont à pondérer par l’existence
de facteurs de risque hémorragique. En curatif, la transfusion de plaquettes est indiquée en cas de saignement actif lorsque la thrombopénie est considérée comme la cause du saignement. En prophylactique, la transfusion de plaquettes doit être prescrite chez l’adulte lorsque le taux est inférieur à 10 G/L, seuil considéré comme à risque hémorragique. Des seuils plus élevés seront à retenir en fonction du contexte clinique (par exemple : thrombopénie centrale profonde avec fièvre, seuil à 20 G/L). Par ailleurs, la Haute Autorité de santé définit des seuils à visée prophylactique en cas de chirurgie ou de geste invasif (par exemple : transfusion si thrombopénie < 50 G/L pour un geste tel que la biopsie ostéomédullaire ou seuil à 100 G/L pour chirurgie ophtalmologique).[4]
L’efficacité entre un concentré plaquettaire d’aphérèse et un mélange de concentrés de plaquettes issus de sang total est identique.
Indications de la transfusion de plasma
- hémorragie d’intensité modérée, peu évolutive ou contrôlée (temps de céphaline activée [TCA] >1,5) ;
- choc hémorragique et situations à risque d’hémorragie massive, en association à des concentrés de globules rouges, avec un ratio plasma frais congelé/concentré de globules rouges compris entre 1/2 et 1/1 ;
- micro-angiopathie thrombotique (purpura thrombotique thrombocytopénique et syndrome hémolytique et urémique avec critères de gravité) ;
- coagulation intravasculaire disséminée obstétricale, lorsque le traitement étiologique ne permet pas de contrôler rapidement l’hémorragie ; coagulation intravasculaire disséminée avec effondrement des facteurs de la coagulation (taux de prothrombine [TP] inférieur à 35-40 %), associée à une hémorragie active ou potentielle (acte invasif) ;
- en cas de surdosage grave en antivitamine-K (AVK), dans deux rares situations : absence de concentrés de complexe prothrombinique, ou absence de concentré de complexe prothombinique ne contenant pas d’héparine en cas d’antécédents de thrombopénie induite par l’héparine (TIH).[5]
Indication des concentrés de granulocytes
Comment prescrire ?
Enfin, il doit figurer sur la prescription de produits sanguins labiles la date et l’heure prévue de la transfusion, ainsi que le degré d’urgence s’il y a lieu.
Trois degrés d’urgence sont individualisés : l’urgence vitale immédiate (délivrance sans délai), l’urgence vitale (délivrance < 30 min) et l’urgence relative (délai de 2 à 3 h).
Analyses immuno-hématologiques et choix des produits
Les analyses obligatoires avant transfusion de concentrés de globules rouges sont le typage ABO-D et le phénotype RH-K du patient réalisés sur deux déterminations, c’est-à-dire sur deux prélèvements issus de deux actes de prélèvement différents et une recherche d’agglutinines irrégulières (RAI) datant de moins de 72 heures. Ce délai peut être étendu à 21 jours en l’absence, certifiée par le prescripteur, d’événements immunisants (grossesse, transfusion) dans les six mois précédents.
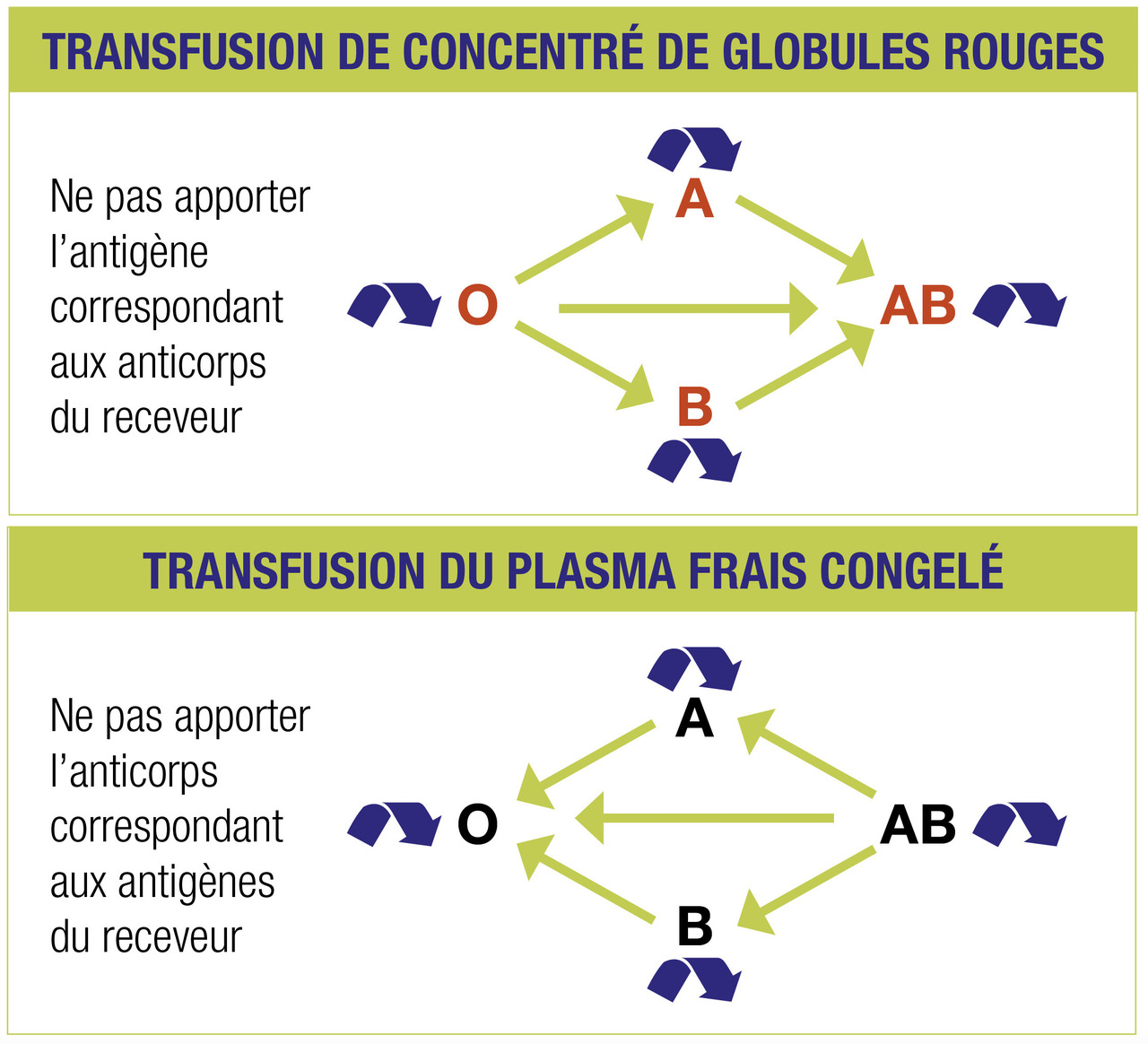
Les modalités de réalisation des examens d’immuno-hématologie sont décrites dans l’arrêté du 15 mai 2018.9 Leurs résultats ainsi que le contexte clinique vont déterminer le choix du produit. Pour tous les patients, la compatibilité ABO-D est respectée (
Le phénotype RH-K comprenant les antigènes C (RH2), E (RH3), c (RH4), e (RH5) et Kell (KEL1) est respecté chez les femmes en âge de procréer (< 50 ans), chez les polytransfusés (hémoglobinopathies…) ainsi que chez les patients possédant une recherche ou un antécédent de recherche d’agglutinines irrégulières positive. On parle de qualification « concentrés de globules rouges phénotypés ».
Le phénotype étendu comprenant les antigènes Jka (JK1), Jkb (JK2), Fya (FY1), Fyb (FY2), S (MNS3), s (MNS4) est respecté lorsqu’à la recherche d’agglutinines irrégulières un allo-anticorps dirigé contre l’un de ces antigènes est mis en évidence.
Une qualification « compatibilisée » est obligatoire en cas de recherche d’agglutinines irrégulières positive ou antécédent de recherche d’agglutinines irrégulières positive et est recommandée chez les patients drépanocytaires. Il s’agit d'une épreuve directe de compatibilité au laboratoire entre le plasma du patient et les globules du concentré de globules rouges sélectionné. Pour une transfusion de plaquettes ou de plasma, il est nécessaire d’avoir deux déterminations de groupage ABO-D.
Certaines situations cliniques nécessitent d’autres qualifications comme la déplasmatisation pour les concentrés de globules rouges et plaquettes en cas d’antécédents allergiques graves et récurrents ou l’irradiation pour les concentrés de globules rouges afin de prévenir la réaction du greffon contre l’hôte chez les patients immmunodéprimés. Depuis que les plaquettes sont exposées aux UVA en présence d’amotosalène, l’irradiation sur celles-ci n’est plus nécessaire.
L’acte transfusionnel
Une fois ces contrôles effectués, la transfusion peut être réalisée. Elle doit bénéficier d’une surveillance particulièrement attentive et continue durant les 15 premières minutes, puis régulière par la suite, à la recherche d’effets indésirables receveurs. Tout professionnel de santé qui est témoin ou qui a connaissance d’un effet indésirable ou d’un incident grave a l’obligation de le signaler sans délai au correspondant d’hémovigilance de l’établissement de santé ou de l’établissement de transfusion sanguine concerné. La traçabilité des produits sanguins labiles transfusés doit être réalisée dans le dossier transfusionnel du patient.
Le système d’hémovigilance
- niveau national : Agence nationale de sécurité du médicament et des produits de santé (ANSM), Santé publique France (ex-InVS), Établissement français du sang, services centraux ;
- niveau régional : coordonnateurs régionaux d’hémovigilance et de sécurité transfusionnelle, correspondants régionaux d’hémovigilance et de sécurité transfusionnelle des établissements de transfusion sanguine (ETS) ;
- niveau local : correspondants d’hémovigilance et de sécurité transfusionnelle des établissements de transfusion sanguine, correspondants d’hémovigilance et de sécurité transfusionnelle des établissements de santé (ES), tout professionnel de santé.
Conduite à tenir immédiate en cas de transfusion mal tolérée
Les effets indésirables receveurs immédiats (< 8 jours post-transfusion)
Réactions immunologiques
Incompatibilité (apport d’anticorps anti-HLA ou HNA) ou activation granulocytaire : ce type d’incident peut être à l’origine d’un œdème pulmonaire lésionnel nommé TRALI (transfusion-related acute lung injury).
Incompatibilité protéique ou autres allergènes (allergie) : les manifestations sont le plus souvent bénignes (urticaire, prurit) mais peuvent dans de très rares cas aboutir à un choc anaphylactique. Ces patients peuvent bénéficier d’une prophylaxie avant transfusion, voire d’une déplasmatisation des concentrés de globules rouges et plaquettes pour les cas les plus sévères.
Incompatibilité plaquettaire : la présence d’anticorps anti-HLA ou anti-HPA chez des patients polytransfusés peuvent aboutir à une inefficacité transfusionnelle plaquettaire,voire un état réfractaire ; des transfusions en concentré de plaquettes d’aphérèse « HLA typés » ou « HPA typés » sont alors indiquées.
Réactions infectieuses : contamination bactérienne
Réactions de surcharge volémique
Effets indésirables receveurs retardés (> 8 jours post-transfusion)
- immunologiques : incompatibilité érythrocytaire (allo-immunisation ou hémolyse retardée), lymphocytaire (GVH) ou plaquettaire (purpura post-transfusionnel) ;
- infectieux : transmission d’agents infectieux conventionnels et non conventionnels ;
- surcharge en fer, notamment chez les polytransfusés (syndrome myélodysplasique ou hémoglobinopathie).
Point sur le risque résiduel infectieux
- lors de l’entretien médical pré-don : un contexte infectieux, des voyages ou des pratiques sexuelles à risque peuvent aboutir à une contre-indication temporaire ou définitive au don. Ceci de manière conforme à l’arrêté relatif aux contre-indications médicales au don de sang ;[10]
- lors de la qualification biologique du don : la recherche de Treponema pallidum et des trois virus VIH, VHB et VHC est obligatoire. Le dépistage des trois virus majeurs se fait par sérologie et par un diagnostic génomique viral, permettant de réduire la fenêtre sérologique silencieuse. En fonction des épidémies saisonnières, d’autres diagnostics génomiques viraux ou des ajournements des donneurs potentiellement exposés sont mis en place (par exemple West Nile, dengue) ;
- lors de la préparation des produits sanguins labiles : les plaquettes et le plasma peuvent subir des techniques d’atténuation des pathogènes par exposition aux UVA en présence d’amotosalène permettant de détruire des virus nus et enveloppés. Concernant le plasma sécurisé par quarantaine, ceux-ci ne seront libérés qu’une fois le contrôle négatif du donneur 60 jours après le don ;
- lors de l’information post-don : le donneur est sensibilisé à signaler à l’EFS l’apparition de signes cliniques après le don, afin de bloquer les produits si nécessaire ou d’informer le prescripteur en cas de produits transfusés. Malgré une prévention importante et l’écoute de la veille sanitaire et épidémiologique, une transmission est possible : chaque année sont publiées les données d’hémovigilance et de sécurité transfusionnelle en France. Le tableau 4 présente les principaux risques transfusionnels (étiologie et prévention), avec les chiffres de risque résiduel correspondant sur l’année 2018.[11]
Médicaments dérivés du sang
L’albumine, les immunoglobulines, certains facteurs de coagulation, les antiprotéases, les colles biologiques sont des médicaments dérivés du plasma sanguin. Ils sont notamment utilisés dans le traitement de déficits immunitaires, des troubles de la coagulation et en réanimation. Comme tout médicament, ils sont soumis à une autorisation de mise sur le marché. Ils ne peuvent être utilisés qu'après évaluation de leur qualité, de leur sécurité et de leur efficacité par l’ANSM. Ils sont également soumis à des règles particulières concernant les modalités de signalement des effets indésirables (pharmacovigilance) et de leur traçabilité.[12] •
POINTS FORTS À RETENIR
Les produits sanguins labiles sont issus d’un don de sang bénévole, anonyme et gratuit. Les principaux sont les concentrés de globules rouges, les concentrés plaquettaires et les plasmas frais congelés. Ils respectent des caractéristiques définies dans un arrêté.
Les indications et les seuils transfusionnels des produits sanguins labiles sont précisés dans les recommandations de la Haute Autorité de santé. Pour la transfusion de concentrés de globules rouges, la tolérance clinique du patient et la présence d’antécédents cardiovasculaires sont les paramètres clés posant l’indication de la transfusion.
Le choix des produits sanguins labiles dépend des résultats des examens d’immuno-hématologie ainsi que du contexte clinique (sexe, âge, pathologie).
La transfusion doit être surveillée, et tout effet indésirable survenant chez un receveur mis en évidence doit être déclaré au correspondant d’hémovigilance de l’établissement de soins. On distingue des effets indésirables immédiats (d’origine immunologique, infectieuse ou de surcharge) et retardés (immunonologique, par exemple allo-immunisation érythrocytaire, non immunologique, par exemple infectieux).
L’hémovigilance est une organisation permettant de surveiller et d’évaluer les effets indésirables des produits sanguins labiles, des effets indésirables des donneurs de sang, des informations post-don et tout incident grave de la chaîne transfusionnelle.
2. Décision du 10 juillet 2018 définissant les principes de bonnes pratiques prévus à l’article L.1222-12 du code de la santé publique.
3. HAS-ANSM. Transfusion de globules rouges homologues : produits, indications, alternatives. Méthode-Recommandations pour la pratique clinique. Novembre 2014.
4. HAS-ANSM. Transfusion de plaquettes : produits, indications. Méthode-Recommandations pour la pratique clinique. Octobre 2015.
5. HAS-ANSM. Transfusion de plasma thérapeutique : produits, indications. Actualisation des recommandations, 2012.
5. Habibi A, et al. Delayed hemolytic transfusion reaction in adult sickle-cell disease: presentations, outcomes, and treatments of 99 referral center episodes. American Journal of Hematology 2016 Oct;91(10):989-94.
6. Agence française de sécurité sanitaire des produits de santé, juin 2003. Transfusion de granulocytes : produits, indications.
7. Circulaire DGS-DHOS-AFSSAPS n° 03-582 du 15 décembre 2003 relative à la réalisation de l’acte transfusionnel.
8. Arrêté du 15 mai 2018 fixant les conditions de réalisation des examens de biologie médicale d’immuno-hématologie érythrocytaire (JORF n° 0116 du 23 mai 2018).
9. Arrêté du 5 avril 2016 fixant les critères de sélection des donneurs de sang.
10. Rapport d’activité d’hémovigilance 2018. https://ansm.sante.fr/S-informer/Points-d-information-Points-d-information/L-ANSM-publie-le-rapport-d-activite-hemovigilance-2018
11. ANSM. https://www.ansm.sante.fr/Dossiers/Medicaments-derives-du-sang/ Situation-des-approvisionnements2/(offset)/0
Dans cet article
- Introduction
- Types de produits sanguins labiles et leurs indications (tableaux 1 et 2)
- Comment prescrire ?
- Analyses immuno-hématologiques et choix des produits
- L’acte transfusionnel
- Le système d’hémovigilance
- Conduite à tenir immédiate en cas de transfusion mal tolérée
- Les effets indésirables receveurs immédiats (< 8 jours post-transfusion)
- Effets indésirables receveurs retardés (> 8 jours post-transfusion)
- Médicaments dérivés du sang
 Encadrés
Encadrés