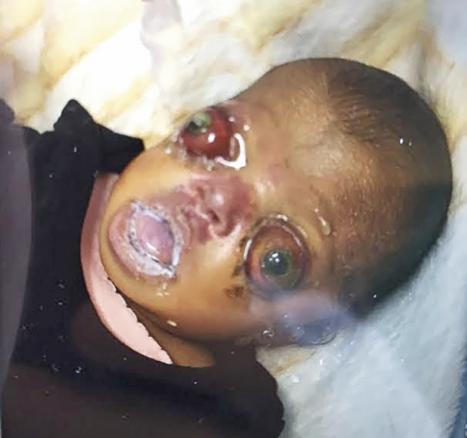
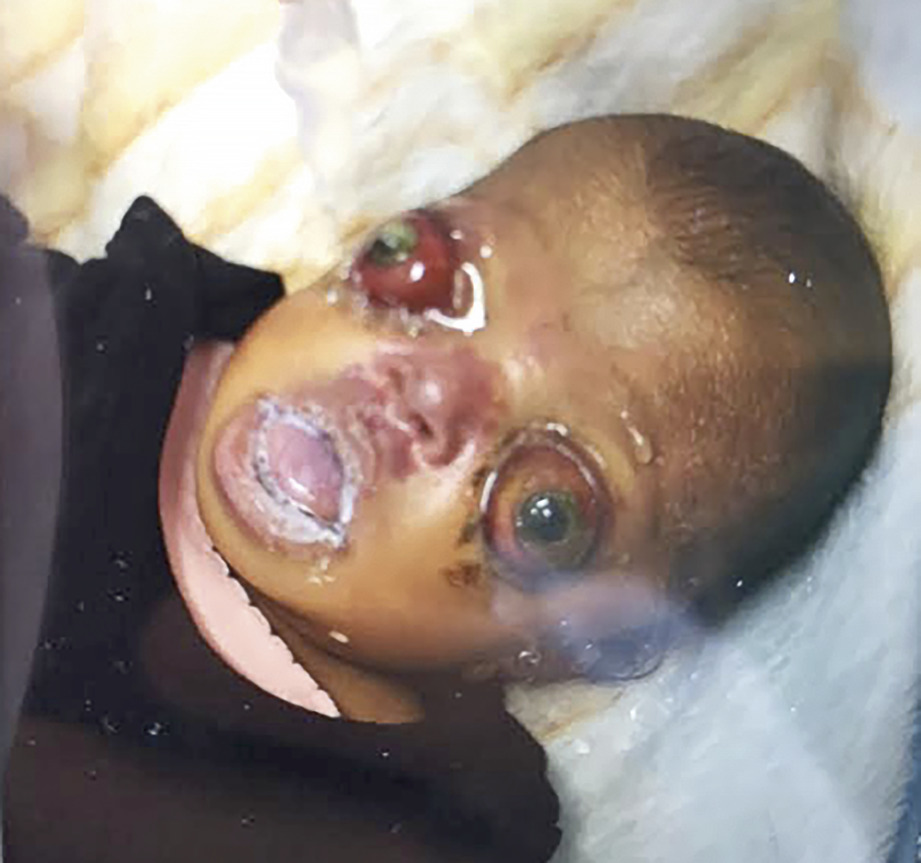
Un enfant de 7 jours (2 kg) est amené en consultation (Mayotte) pour une exophtalmie impressionnante évoquant d’emblée un syndrome de Crouzon.
Il est fébrile (38,2 °C), sa fréquence cardiaque est à 186/min et sa pression artérielle à 68/33 mmHg. L’examen montre une cornée sèche et opaque, un rétrognatisme, une macroglossie, une arthrogrypose des coudes et des genoux.
Au bilan biologique : glycémie à 3,65 g/L, cholestase (bilirubine libre : 206 µmol/L), acidose respiratoire (pH 7,17).
Il est fébrile (38,2 °C), sa fréquence cardiaque est à 186/min et sa pression artérielle à 68/33 mmHg. L’examen montre une cornée sèche et opaque, un rétrognatisme, une macroglossie, une arthrogrypose des coudes et des genoux.
Au bilan biologique : glycémie à 3,65 g/L, cholestase (bilirubine libre : 206 µmol/L), acidose respiratoire (pH 7,17).
Les craniosynostoses, ou craniosténoses, dues à la fermeture prématurée d’une ou plusieurs sutures de la voûte crânienne, ont une incidence d’environ 1 cas sur 2 500 naissances. Le trouble de croissance du crâne, du cerveau et du système nerveux est responsable de déformations morphologiques crâniennes, du visage et de la mâchoire, parfois de troubles respiratoires et de cécité.
La forme la plus fréquente est la dystocie cranio- faciale, ou syndrome de Crouzon, maladie génétique rare (1 cas sur 60 000 naissances) à transmission autosomique dominante. Décrite en 1912,1 elle associe une craniosynostose et une dysmorphie faciale : hypertélorisme (accroissement de la distance séparant les 2 orbites), hypoplasie importante du maxillaire supérieur, convexité du nez, implantation basse des oreilles, prognathisme et exorbitisme majeur. Un acanthosis nigricans est parfois retrouvé. Le degré des malformations est variable, selon le nombre et la position des sutures concernées.
Le blocage de la croissance du crâne provoque une hypertension intracrânienne, avec hydrocéphalie (30 % des cas) et compression des nerfs auditifs et optiques responsables d’une surdité (55 % des cas) et d’une cécité. Une déficience mentale est souvent constatée. L’exorbitisme est dû à la réduction de la capacité orbitaire, rendant impossible l’occlusion des yeux. L’hypertélorisme est lié aux sutures prématurées sphénozygomatiques et sphénotemporales. L’obstruction des voies aériennes supérieures peut entraîner des troubles respiratoires.2
L’imagerie du crâne confirme la disparition des sutures crâniennes, l’élargissement de la fosse hypophysaire, l’hypo- plasie maxillaire et le manque de profondeur des orbites ; souvent, on retrouve une fusion des vertèbres cervicales C2 et C3. Un scanner tridimensionnel peut objectiver des impressions digitiformes de la voûte.
Dans certains cas, le diagnostic est évoqué à l’échographie anténatale, montrant les sutures précoces des os du crâne.3 L’analyse génétique retrouve une mutation au niveau du récepteur 2 du fibroblast growth factor (FGFR2, locus q26 du chromosome 10). 70 % des cas ont un caractère familial.4 Le diagnostic différentiel doit être fait avec d’autres cranio- synostoses comme les syndromes d’Apert (syndactylie des mains et des pieds : accolement ou fusion complète de 2 ou plusieurs doigts) et de Pfeiffer (ostéochondrodysplasie).5
Dans les cas modérés, des traitements symptomatiques peuvent être tentés : dérivation ventriculo-péritonéale contre l’hypertension intracrânienne, tarsorraphie en cas d’exorbitisme, trachéotomie si insuffisance respiratoire, chirurgie réparatrice maxillo-faciale de l’hypoplasie du maxillaire et des orbites. Toutefois, les complications sont nombreuses. Chez ce nourrisson, après plusieurs épisodes de bradycardie et de désaturation, l’évolution a été rapidement fatale.
La forme la plus fréquente est la dystocie cranio- faciale, ou syndrome de Crouzon, maladie génétique rare (1 cas sur 60 000 naissances) à transmission autosomique dominante. Décrite en 1912,1 elle associe une craniosynostose et une dysmorphie faciale : hypertélorisme (accroissement de la distance séparant les 2 orbites), hypoplasie importante du maxillaire supérieur, convexité du nez, implantation basse des oreilles, prognathisme et exorbitisme majeur. Un acanthosis nigricans est parfois retrouvé. Le degré des malformations est variable, selon le nombre et la position des sutures concernées.
Le blocage de la croissance du crâne provoque une hypertension intracrânienne, avec hydrocéphalie (30 % des cas) et compression des nerfs auditifs et optiques responsables d’une surdité (55 % des cas) et d’une cécité. Une déficience mentale est souvent constatée. L’exorbitisme est dû à la réduction de la capacité orbitaire, rendant impossible l’occlusion des yeux. L’hypertélorisme est lié aux sutures prématurées sphénozygomatiques et sphénotemporales. L’obstruction des voies aériennes supérieures peut entraîner des troubles respiratoires.2
L’imagerie du crâne confirme la disparition des sutures crâniennes, l’élargissement de la fosse hypophysaire, l’hypo- plasie maxillaire et le manque de profondeur des orbites ; souvent, on retrouve une fusion des vertèbres cervicales C2 et C3. Un scanner tridimensionnel peut objectiver des impressions digitiformes de la voûte.
Dans certains cas, le diagnostic est évoqué à l’échographie anténatale, montrant les sutures précoces des os du crâne.3 L’analyse génétique retrouve une mutation au niveau du récepteur 2 du fibroblast growth factor (FGFR2, locus q26 du chromosome 10). 70 % des cas ont un caractère familial.4 Le diagnostic différentiel doit être fait avec d’autres cranio- synostoses comme les syndromes d’Apert (syndactylie des mains et des pieds : accolement ou fusion complète de 2 ou plusieurs doigts) et de Pfeiffer (ostéochondrodysplasie).5
Dans les cas modérés, des traitements symptomatiques peuvent être tentés : dérivation ventriculo-péritonéale contre l’hypertension intracrânienne, tarsorraphie en cas d’exorbitisme, trachéotomie si insuffisance respiratoire, chirurgie réparatrice maxillo-faciale de l’hypoplasie du maxillaire et des orbites. Toutefois, les complications sont nombreuses. Chez ce nourrisson, après plusieurs épisodes de bradycardie et de désaturation, l’évolution a été rapidement fatale.
Références
1. Crouzon LEO. Dysostose cranio-faciale héréditaire. Bull Soc Med Hôp Paris 1912;33:545-55.
2. Mathijssen IM. Guideline for care of patients with the diagnoses of cranio- synostosis. Working Group on Craniosynostosis. J Craniofac Surg 2015;26:1735-807.
3. Renier D, Lajeunie E, Catala M, et al. Craniosténoses. EMC (Elsevier Masson SAS) - Pédiatrie-Maladies infectieuses;2008:1-19 [Article 4-096-B-10].
4. Al-Namnam NM, Hariri F, Thong MK, Rahman ZA. Crouzon syndrome: genetic and intervention review. J Oral Biol Craniofac Res 2019;9:37-9.
5. Armand T, Schaefer E, Di Rocco F, Edery P, Collet C, Rossi M. Genetic bases of craniosynostoses: an update. Neurochirurgie 2019;65:196-201.
2. Mathijssen IM. Guideline for care of patients with the diagnoses of cranio- synostosis. Working Group on Craniosynostosis. J Craniofac Surg 2015;26:1735-807.
3. Renier D, Lajeunie E, Catala M, et al. Craniosténoses. EMC (Elsevier Masson SAS) - Pédiatrie-Maladies infectieuses;2008:1-19 [Article 4-096-B-10].
4. Al-Namnam NM, Hariri F, Thong MK, Rahman ZA. Crouzon syndrome: genetic and intervention review. J Oral Biol Craniofac Res 2019;9:37-9.
5. Armand T, Schaefer E, Di Rocco F, Edery P, Collet C, Rossi M. Genetic bases of craniosynostoses: an update. Neurochirurgie 2019;65:196-201.
Une question, un commentaire ?