La vascularite à IgA se caractérise par une atteinte inflammatoire avec dépôts d’IgA dans les vaisseaux de petit calibre. Elle évolue par poussées et se manifeste par des atteintes cutanée (purpura vasculaire), articulaire, digestive et/ou rénale. Son pronostic est lié aux atteintes gastro-intestinales et aux éventuelles lésions rénales. Sa prise en charge dans les formes sévères repose sur l’administration de corticoïdes.
La vascularite à immunoglobulines A (VIgA), anciennement appelée purpura rhumatoïde ou syndrome d’Henoch-Schönlein, est une vascularite systémique définie par une inflammation des vaisseaux sanguins conduisant à une altération de la paroi vasculaire. Elle touche les vaisseaux de petit calibre (artérioles, capillaires et veinules).1 La VIgA touche plus souvent les enfants que les adultes. Elle peut se manifester par une atteinte cutanée, articulaire et gastro-intestinale et/ou rénale. Cette vascularite évolue par poussées et peut guérir spontanément. Son pronostic à court terme est lié à la gravité d’une éventuelle atteinte aiguë gastro-intestinale (risque de perforation digestive), tandis que son pronostic à long terme dépend des éventuelles lésions rénales (risque d’insuffisance rénale chronique).
La VIgA est secondaire à la formation de complexes immuns à partir d’un sous-type d’immunoglobulines A (IgA1) se déposant au sein des vaisseaux de petit calibre. Ce phénomène pourrait résulter d’une synthèse accrue, d’un défaut de clairance ou d’une anomalie structurelle des IgA.2 L’IgA est la classe d’immunoglobulines la plus importante dans l’immunité des muqueuses. Il existe donc un lien étroit entre cette interface et de potentiels facteurs déclenchants à localisation muqueuse (infections, néoplasies…). Un terrain favorisant associé à un événement déclenchant identifié ou non pourrait être à l’origine de l’apparition de la maladie (cirrhose, maladie inflammatoire du tube digestif, spondylarthrite ankylosante…) [tableau 1 ].
La VIgA est secondaire à la formation de complexes immuns à partir d’un sous-type d’immunoglobulines A (IgA1) se déposant au sein des vaisseaux de petit calibre. Ce phénomène pourrait résulter d’une synthèse accrue, d’un défaut de clairance ou d’une anomalie structurelle des IgA.2 L’IgA est la classe d’immunoglobulines la plus importante dans l’immunité des muqueuses. Il existe donc un lien étroit entre cette interface et de potentiels facteurs déclenchants à localisation muqueuse (infections, néoplasies…). Un terrain favorisant associé à un événement déclenchant identifié ou non pourrait être à l’origine de l’apparition de la maladie (cirrhose, maladie inflammatoire du tube digestif, spondylarthrite ankylosante…) [
Le purpura, un signe clinique constant
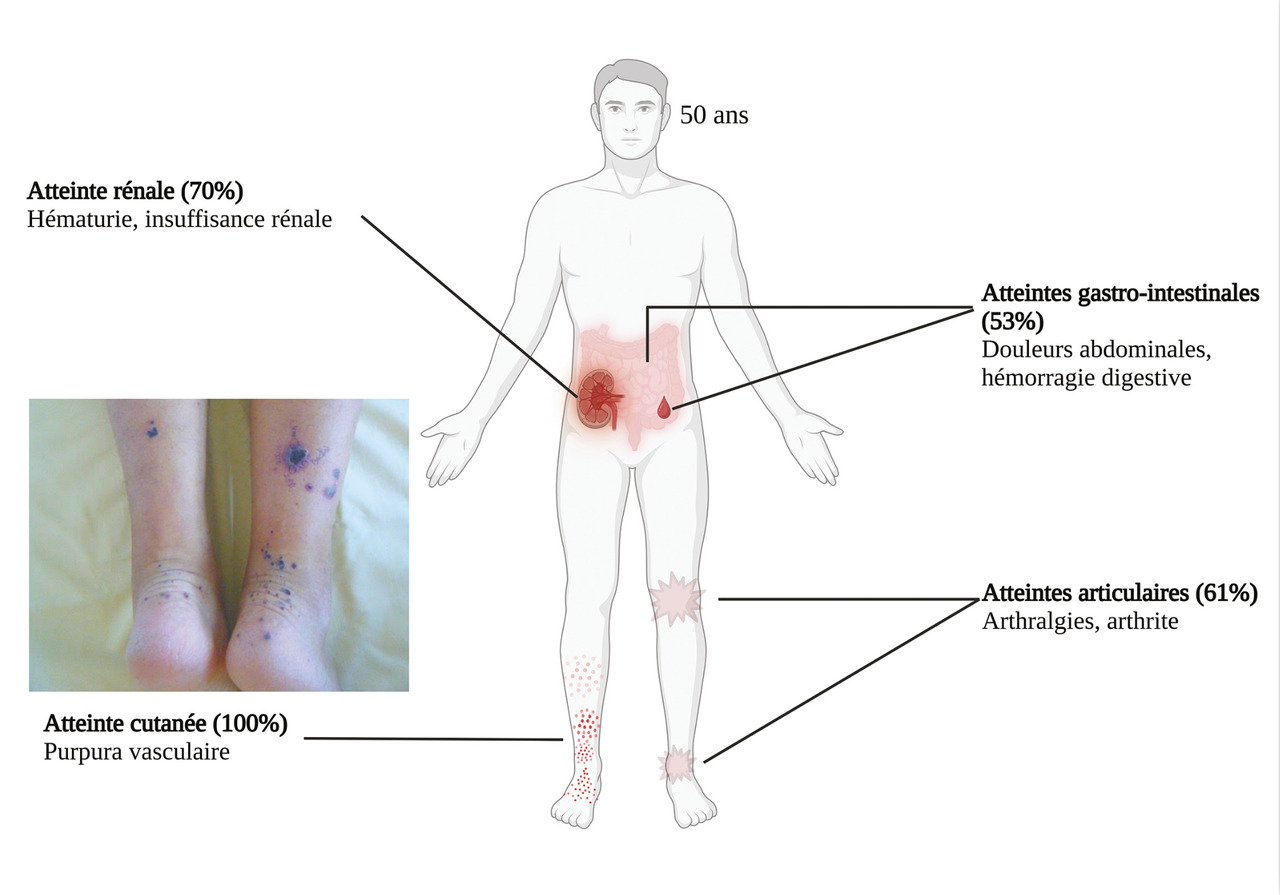
Les principales manifestations cliniques des VIgA sont représentées sur la figure 1 .3
Un purpura symétrique et ascendant
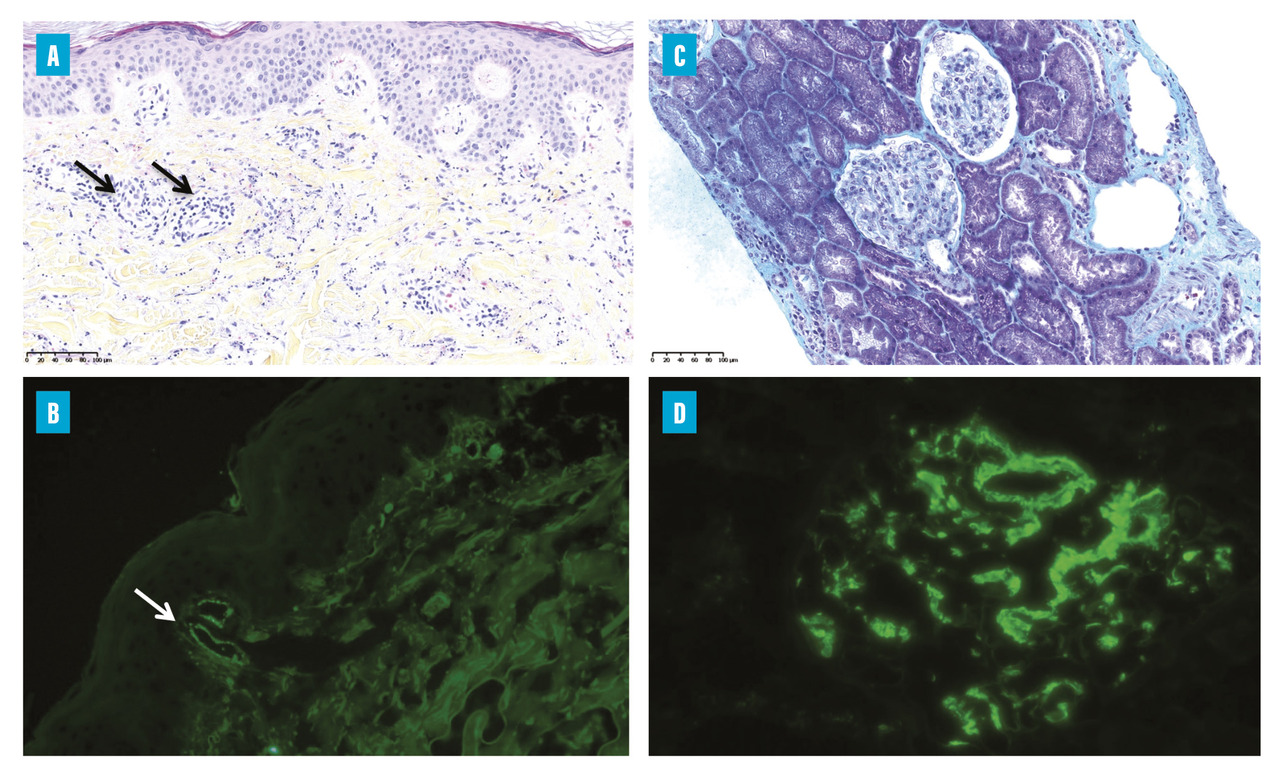
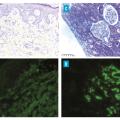
La manifestation clinique initiale constante est un purpura déclive, pétéchial ou ecchymotique, voire nécrotique (fig. 1 ). Ces lésions sont symétriques, prédominent aux zones de pression mais peuvent s’étendre à l’ensemble du tégument (cutané ou muqueux) dans certaines formes cutanées sévères ; elles ont une évolution ascendante. Le purpura évolue par poussées régressant spontanément en quinze jours. L’analyse histologique d’une biopsie cutanée permet de confirmer le diagnostic en objectivant un infiltrat inflammatoire associé à une fragmentation de polynucléaires neutrophiles, avec ou sans nécrose caractéristique d’une vascularite leucocytoclasique, avec parfois en immunofluorescence des dépôts d’IgA au niveau de la paroi des vaisseaux, surtout à la phase aiguë du purpura (fig. 2 ). L’immunofluorescence peut revenir positive dans environ 60 % des cas.4
Des arthralgies surtout présentes aux membres inférieurs
Les manifestations articulaires sont présentes dans deux tiers des cas et concernent le plus souvent plusieurs articulations. Elles se manifestent fréquemment sous la forme d’arthralgies et très rarement sous forme d’arthrites non destructrices touchant préférentiellement les genoux et chevilles. Peu intenses, elles sont rapidement résolutives en deux semaines.
Une atteinte digestive présente dans un cas sur deux
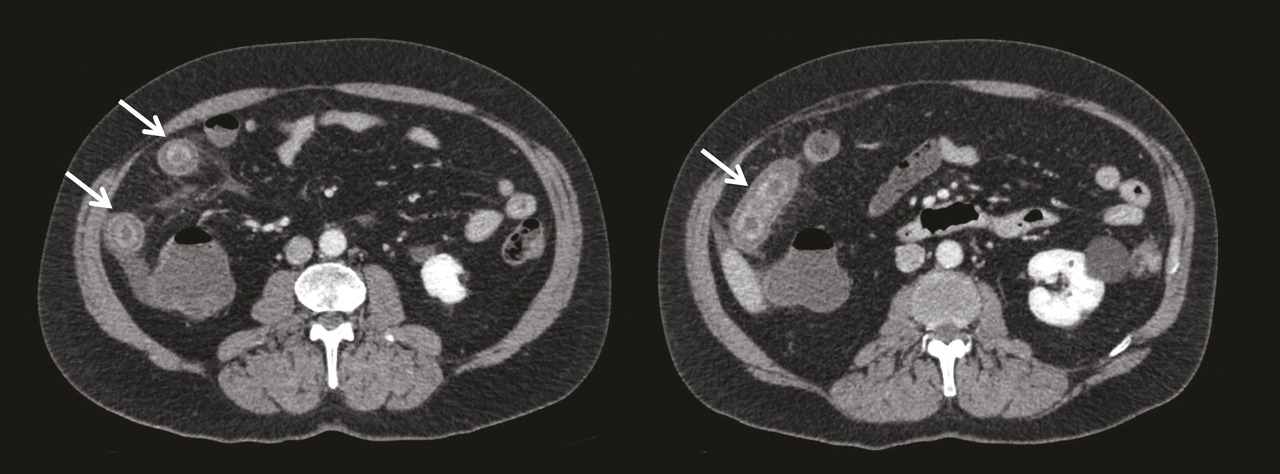
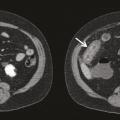
L’atteinte digestive est également fréquente et touche plus de la moitié des patients. Elle se manifeste généralement dans la semaine suivant l’apparition du purpura avec des douleurs abdominales épigastriques par atteinte duodénale, ou en regard de la fosse iliaque en cas d’iléite. Un saignement digestif peut survenir, chez 31 % des patients présentant une atteinte digestive, et être parfois gravissime, engageant le pronostic vital. Cependant, il est le plus souvent occulte ou sous forme de méléna. L’échographie ou le scanner peut être utile, montrant une paroi intestinale épaissie (fig. 3 ). La réalisation d’endoscopies digestive haute et basse permet d’objectiver un purpura muqueux, mais son intérêt est controversé du fait d’un risque de perforation digestive.
L’atteinte rénale impacte le pronostic
L’atteinte rénale survient dans plus de la moitié des cas,3 au diagnostic principalement mais aussi au cours des premiers mois d’évolution de la maladie. Elle impacte le pronostic à long terme en raison du risque d’insuffisance rénale chronique. Le signe le plus précoce est une hématurie microscopique (supérieure à 10 hématies/mm3). D’autres signes peuvent s’y associer comme une hypertension artérielle, une protéinurie d’origine glomérulaire de rang variable et une insuffisance rénale. L’insuffisance rénale est beaucoup plus fréquente chez l’adulte que chez l’enfant, avec une incidence allant jusqu’à 30 % selon les séries.5 La biopsie rénale et les résultats histologiques orientent la prise en charge thérapeutique et ont une valeur pronostique. On y retrouve une néphropathie glomérulaire avec dépôts d’IgA en immunofluorescence (fig. 2 ). La néphropathie de la vascularite à IgA est identique à la néphropathie à IgA (anciennement maladie de Berger), et seul le contexte clinique avec des éléments extrarénaux permet de les distinguer. Ces dépôts d’IgA sont localisés dans le mésangium de tous les glomérules rénaux accompagnés parfois d’une prolifération cellulaire glomérulaire. Une classification a été proposée chez l’adulte, et la classe la plus fréquemment retrouvée était la glomérulonéphrite endocapillaire diffuse.6
Des signes généraux peu spécifiques
Enfin, des signes généraux modérés sont fréquemment retrouvés (fièvre, asthénie, perte de poids) alors que d’autres atteintes sont beaucoup plus rares, comme l’atteinte neurologique périphérique (multinévrite), testiculaire (orchite), pulmonaire (hémorragie intra-alvéolaire).
Une longue liste de facteurs favorisants
La VIgA peut se manifester à tout âge, mais il existe deux pics d’incidence de la maladie : un premier chez l’enfant entre 3 et 15 ans, puis un second chez l’adulte à 50 ans, avec une prédominance masculine.7
L’incidence augmente en hiver en lien avec les infections saisonnières, notamment de la sphère otorhinolaryngologique et du tractus respiratoire.8 Une centaine d’agents pathogènes ont été incriminés. Les plus souvent retrouvés comme potentiels déclencheurs de VIgA sont les streptocoques, Helicobacter pylori, Mycoplasma pneumoniae, le parvovirus B19 et le virus de l’hépatite A.9 Certaines études ont souligné un possible lien entre infection à severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) et VIgA.10 Durant la pandémie, une diminution de fréquence des VIgA de l’adulte a été observée, probablement en raison du port du masque généralisé qui a permis une diminution de la circulation des virus respiratoires.11
Il existe aussi des facteurs déclenchants non infectieux, comme certaines prises médicamenteuses telles que des antibiotiques de la famille des pénicillines ou encore des biothérapies ciblant le tumor necrosis factor alpha (TNF α). Des études ont mentionné la survenue de VIgA et de rechute de la maladie après vaccination anti-SARS-CoV-2.12
Enfin, il existe une association entre la VIgA et certains cancers, touchant notamment les voies aéro-digestives, le tractus urinaire mais également les cancers glandulaires comme le cancer du sein.13 Leur présentation initiale et leur évolution sembleraient plus sévères, et toucheraient principalement des patients de plus de 50 ans. En termes chronologiques, le cancer est majoritairement diagnostiqué dans les suites des explorations réalisées dans le cadre du bilan de la vascularite.
Des cas de VIgA survenant au cours de pathologies lymphoïdes telles qu’une gammapathie monoclonale à IgA ou qu’un myélome à IgA ont aussi été décrits. Tout comme les cancers solides, leur présentation semble plus sévère, avec une plus forte résistance aux traitements.14
Enfin, la cirrhose, notamment d’origine alcoolique, est un terrain pouvant favoriser la survenue de VIgA. Dans ce cas, la vascularite n’est pas plus sévère, et la mortalité est essentiellement grevée par la sévérité de la cirrhose.15 Les différents terrains et facteurs favorisants sont indiqués dans letableau 1 .
L’incidence augmente en hiver en lien avec les infections saisonnières, notamment de la sphère otorhinolaryngologique et du tractus respiratoire.8 Une centaine d’agents pathogènes ont été incriminés. Les plus souvent retrouvés comme potentiels déclencheurs de VIgA sont les streptocoques, Helicobacter pylori, Mycoplasma pneumoniae, le parvovirus B19 et le virus de l’hépatite A.9 Certaines études ont souligné un possible lien entre infection à severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) et VIgA.10 Durant la pandémie, une diminution de fréquence des VIgA de l’adulte a été observée, probablement en raison du port du masque généralisé qui a permis une diminution de la circulation des virus respiratoires.11
Il existe aussi des facteurs déclenchants non infectieux, comme certaines prises médicamenteuses telles que des antibiotiques de la famille des pénicillines ou encore des biothérapies ciblant le tumor necrosis factor alpha (TNF α). Des études ont mentionné la survenue de VIgA et de rechute de la maladie après vaccination anti-SARS-CoV-2.12
Enfin, il existe une association entre la VIgA et certains cancers, touchant notamment les voies aéro-digestives, le tractus urinaire mais également les cancers glandulaires comme le cancer du sein.13 Leur présentation initiale et leur évolution sembleraient plus sévères, et toucheraient principalement des patients de plus de 50 ans. En termes chronologiques, le cancer est majoritairement diagnostiqué dans les suites des explorations réalisées dans le cadre du bilan de la vascularite.
Des cas de VIgA survenant au cours de pathologies lymphoïdes telles qu’une gammapathie monoclonale à IgA ou qu’un myélome à IgA ont aussi été décrits. Tout comme les cancers solides, leur présentation semble plus sévère, avec une plus forte résistance aux traitements.14
Enfin, la cirrhose, notamment d’origine alcoolique, est un terrain pouvant favoriser la survenue de VIgA. Dans ce cas, la vascularite n’est pas plus sévère, et la mortalité est essentiellement grevée par la sévérité de la cirrhose.15 Les différents terrains et facteurs favorisants sont indiqués dans le
Écarter les diagnostics différentiels
Le diagnostic de VIgA repose sur l’association de signes cliniques évocateurs associés à des résultats anatomopathologiques et sur l’élimination des diagnostics différentiels.
Comme décrit précédemment, la VIgA doit être évoquée devant la présence d’un purpura vasculaire cutané associé à des manifestations articulaires, digestives ou rénales. L’histologie est très utile au diagnostic, surtout si des dépôts d’IgA sont mis en évidence au niveau cutané (vascularite leucocytoclasique) et/ou rénal (néphropathie glomérulaire) [fig. 2 ]. Aucun signe biologique n’est spécifique de la maladie. L’élévation isolée des IgA sériques n’est observée que dans 60 % des cas et peut orienter vers ce diagnostic. Le syndrome inflammatoire est généralement peu marqué, sauf en cas d’atteinte digestive sévère ou d’infection concomitante.
L’élimination des diagnostics différentiels est essentielle. Il convient d’abord d’écarter les autres causes de purpura : une thrombopénie, une hémopathie ou une maladie infectieuse bactérienne (purpura fulminans, endocardite infectieuse) ou virale (virus de l’immunodéficience humaine [VIH], hépatites virales). D’autres types de vascularites primitives et secondaires doivent également être éliminés, comme la vascularite associée aux anticorps anticytoplasme des polynucléaires neutrophiles (ANCA) et la vascularite cryoglobulinémique. La recherche d’une pathologie néoplasique sous-jacente, avec des examens adaptés, peut également se justifier en cas de survenue d’une VIgA après 50 ans.
Il existe des critères de classification chez l’enfant, qui sont extrapolés mais non validés chez l’adulte, ce qui en complique l’interprétation.16 Les différents examens paracliniques utiles à la recherche de diagnostics différentiels sont détaillés dans letableau 2 .
Comme décrit précédemment, la VIgA doit être évoquée devant la présence d’un purpura vasculaire cutané associé à des manifestations articulaires, digestives ou rénales. L’histologie est très utile au diagnostic, surtout si des dépôts d’IgA sont mis en évidence au niveau cutané (vascularite leucocytoclasique) et/ou rénal (néphropathie glomérulaire) [
L’élimination des diagnostics différentiels est essentielle. Il convient d’abord d’écarter les autres causes de purpura : une thrombopénie, une hémopathie ou une maladie infectieuse bactérienne (purpura fulminans, endocardite infectieuse) ou virale (virus de l’immunodéficience humaine [VIH], hépatites virales). D’autres types de vascularites primitives et secondaires doivent également être éliminés, comme la vascularite associée aux anticorps anticytoplasme des polynucléaires neutrophiles (ANCA) et la vascularite cryoglobulinémique. La recherche d’une pathologie néoplasique sous-jacente, avec des examens adaptés, peut également se justifier en cas de survenue d’une VIgA après 50 ans.
Il existe des critères de classification chez l’enfant, qui sont extrapolés mais non validés chez l’adulte, ce qui en complique l’interprétation.16 Les différents examens paracliniques utiles à la recherche de diagnostics différentiels sont détaillés dans le
Évolution et pronostic
L’évolution des formes cutanéo-articulaires est habituellement favorable en une quinzaine de jours. Cependant, près d’un quart des patients présentent plusieurs poussées de la maladie et près d’un tiers ont une forme chronique de la maladie sous forme de glomérulonéphrite chronique.
Le pronostic à court terme est essentiellement lié à la sévérité de l’atteinte digestive, notamment en cas de syndrome hémorragique ou de perforations digestives, avec une mortalité estimée à 2 %.17
Le pronostic à long terme est lié à l’atteinte rénale. Le risque d’évolution vers une insuffisance rénale chronique est de l’ordre de 18 %.
Le pronostic à court terme est essentiellement lié à la sévérité de l’atteinte digestive, notamment en cas de syndrome hémorragique ou de perforations digestives, avec une mortalité estimée à 2 %.17
Le pronostic à long terme est lié à l’atteinte rénale. Le risque d’évolution vers une insuffisance rénale chronique est de l’ordre de 18 %.
Prise en charge multidisciplinaire, le plus souvent symptomatique
La prise en charge d’un patient présentant une vascularite à IgA doit être multidisciplinaire, en lien avec un médecin hospitalier, un centre de référence ou de compétence ainsi que les correspondants de différentes spécialités (interniste, néphrologue, dermatologue…).
Pour la plupart des patients, des mesures symptomatiques suffisent. Le repos au lit et le port de bas de compression médicale limitent l’extension du purpura sans influer sur l’évolution de l’atteinte digestive. Les douleurs articulaires et musculaires sont sensibles aux antalgiques de paliers I ou II. La prescription d’anti-inflammatoires non stéroïdiens (AINS) est à éviter compte-tenu de leur potentiel effet délétère sur la fonction rénale et leur effet antiagrégant plaquettaire. La colchicine peut être utilisée en cas d’atteinte cutanée ou articulaire récidivante, en tenant compte des risques d’interactions médicamenteuses. En cas d’atteinte rénale, un suivi spécialisé et des mesures de néphroprotection sont indiqués, avec la nécessité de contrôler la pression artérielle et la protéinurie éventuelle, notamment par l’utilisation d’un inhibiteur du système rénine-angiotensine.
En cas de forme clinique plus sévère, des traitements spécifiques peuvent également être utilisés ; corticothérapie orale ou intraveineuse, plus ou moins associée à un autre immunosuppresseur. La dose et la durée de la corticothérapie ne sont pas codifiées, allant de quelques semaines pour l’atteinte digestive à quelques mois en cas d’atteinte rénale. Aucun essai n’a, à ce jour, démontré une efficacité, chez l’adulte, d’immunosuppresseurs ou de biothérapies. Cependant, ils doivent systématiquement être discutés avec un spécialiste au cours de manifestations rénales ou digestives sévères.
Pour la plupart des patients, des mesures symptomatiques suffisent. Le repos au lit et le port de bas de compression médicale limitent l’extension du purpura sans influer sur l’évolution de l’atteinte digestive. Les douleurs articulaires et musculaires sont sensibles aux antalgiques de paliers I ou II. La prescription d’anti-inflammatoires non stéroïdiens (AINS) est à éviter compte-tenu de leur potentiel effet délétère sur la fonction rénale et leur effet antiagrégant plaquettaire. La colchicine peut être utilisée en cas d’atteinte cutanée ou articulaire récidivante, en tenant compte des risques d’interactions médicamenteuses. En cas d’atteinte rénale, un suivi spécialisé et des mesures de néphroprotection sont indiqués, avec la nécessité de contrôler la pression artérielle et la protéinurie éventuelle, notamment par l’utilisation d’un inhibiteur du système rénine-angiotensine.
En cas de forme clinique plus sévère, des traitements spécifiques peuvent également être utilisés ; corticothérapie orale ou intraveineuse, plus ou moins associée à un autre immunosuppresseur. La dose et la durée de la corticothérapie ne sont pas codifiées, allant de quelques semaines pour l’atteinte digestive à quelques mois en cas d’atteinte rénale. Aucun essai n’a, à ce jour, démontré une efficacité, chez l’adulte, d’immunosuppresseurs ou de biothérapies. Cependant, ils doivent systématiquement être discutés avec un spécialiste au cours de manifestations rénales ou digestives sévères.
Maladie systémique rare de l’adulte
La vascularite à IgA est une maladie systémique rare de l’adulte. Elle se caractérise par une atteinte inflammatoire avec dépôts d’IgA dans les vaisseaux de petit calibre et se manifeste par des atteintes cutanée, articulaire, digestive et rénale. Le pronostic vital peut être engagé à court terme par une atteinte digestive sévère. Le pronostic à long terme est, quant à lui, lié à l’atteinte rénale. Le diagnostic repose sur un faisceau d’arguments cliniques et histologiques et sur l’élimination des diagnostics différentiels. L’anatomopathologie confirme le diagnostic. La prise en charge est principalement symptomatique (repos, antalgie, mesures hygiéno-diététiques et de néphroprotection). La prise en charge spécifique (hospitalisation, corticothérapie, voire traitement immunosuppresseur) n’est à ce jour pas codifiée. Un groupe européen multidisciplinaire d’experts (EUGAVAS Study Group) s’est récemment formé, dans le but d’émettre des recommandations pour améliorer et harmoniser la prise en charge des malades. La réalisation d’essais randomisés contrôlés dans les années à venir reste un défi du fait de la rareté de la pathologie, et permettrait une avancée significative dans le traitement des patients. ●
Les auteurs remercient le Dr Kervarrec, anatomopathologiste au CHU de Tours, pour la réalisation des photographies de l’histologie cutanée et rénale.
Références
1. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013;65:1-11.
2. Heineke MH, van Egmond M. Immunoglobulin A: Magic bullet or Trojan horse? Eur J Clin Invest 2017;47:184-92.
3. Audemard-Verger A, Terrier B, Dechartres A, Chanal J, Amoura Z, Le Gouellec N, et al. Characteristics and management of IgA vasculitis (Henoch-Schönlein) in adults: Data from 260 patients included in a French multicenter retrospective survey. Arthritis Rheumatol 2017;69:1862-70.
4. Lath K, Chatterjee D, Saikia UN, Saikia B, Minz R, De D, et al. Role of direct immunofluorescence in cutaneous small-vessel vasculitis: Experience from a tertiary center. Am J Dermatopathol 2018;40:661.
5. Berthelot L, Jamin A, Viglietti D, Chemouny JM, Ayari H, Pierre M, et al. Value of biomarkers for predicting immunoglobulin A vasculitis nephritis outcome in an adult prospective cohort. Nephrol Dial Transplant 2018;33(9): 1579-90.
6. Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein Purpura in adults: Outcome and prognostic factors. J Am Soc Nephrol 2002;13:1271-8.
7. Watts RA, Lane S, Scott DGI. What is known about the epidemiology of the vasculitides? Best Pract Res Clin Rheumatol 2005;19:191-207.
8. Maisons V, Ramdani Y, Messiaen C, Sautenet B, Halimi JM, Maillot F, et al. Épidémiologie de la vascularite à IgA de l’enfant et de l’adulte en France à partir de la BNDMR. Rev Medecine Interne 2022;43:A380.
9. Nikolaishvili M, Pazhava A, Di Lernia V. Viral infections may be associated with Henoch–Schönlein Purpura. J Clin Med 2023;12:697.
10. Ramdani Y, Galempoix JM, Augusto JF, Dekmeer E, Perard L, Ferreira N, et al. Immunoglobulin a vasculitis following COVID-19: A French multicenter case series. J Rheumatol 2022;49:1390-4.
11. Batu ED, Sarı A, Erden A, Sönmez HE, Armağan B, Kalyoncu U, et al. Comparing immunoglobulin A vasculitis (Henoch-Schönlein purpura) in children and adults: A single-centre study from Turkey. Scand J Rheumatol 2018;47:481-6.
12. Ramdani Y, Bettuzzi T, Bouznad A, Delaitre L, Nassarmadji K, Didier K, et al. IgA vasculitis following COVID-19 vaccination: A French multicentre case series including 12 patients. J Rheumatol 2023;50(2):252-7.
13. Hankard A, Michot JM, Terrier B, Brihaye B, Chanal J, Combe C, et al. New insights on IgA vasculitis with underlying solid tumor: A nationwide French study of 30 patients. Clin Rheumatol 2021;40:1933-40.
14. Umemura H, Yamasaki O, Iwatsuki K. Leukocytoclastic vasculitis associated with immunoglobulin A lambda monoclonal gammopathy of undetermined significance: A case report and review of previously reported cases. J Dermatol 2018;45:1009-12.
15. Elhani I, Pillebout E, Terrier B, Hankard A, Vrtovsnik F, Jourde-Chiche N, et al. IgA vasculitis with underlying liver cirrhosis: A French nationwide case series of 20 patients. J Rheumatol 2021;48:735.
16. Hočevar A, Rotar Z, Jurčić V, Pižem J, Čučnik S, Vizjak A, et al. IgA vasculitis in adults: The performance of the EULAR/PRINTO/PRES classification criteria in adults. Arthritis Res Ther 2016;18:58.
17. Audemard-Verger A, Pillebout E, Amoura Z, Cacoub P, Jourde-Chiche N, Lioger B, et al. Gastrointestinal involvement in adult IgA vasculitis (Henoch-Schönlein purpura): Updated picture from a French multicentre and retrospective series of 260 cases. Rheumatol Oxf Engl 2020;59:3050-7.
2. Heineke MH, van Egmond M. Immunoglobulin A: Magic bullet or Trojan horse? Eur J Clin Invest 2017;47:184-92.
3. Audemard-Verger A, Terrier B, Dechartres A, Chanal J, Amoura Z, Le Gouellec N, et al. Characteristics and management of IgA vasculitis (Henoch-Schönlein) in adults: Data from 260 patients included in a French multicenter retrospective survey. Arthritis Rheumatol 2017;69:1862-70.
4. Lath K, Chatterjee D, Saikia UN, Saikia B, Minz R, De D, et al. Role of direct immunofluorescence in cutaneous small-vessel vasculitis: Experience from a tertiary center. Am J Dermatopathol 2018;40:661.
5. Berthelot L, Jamin A, Viglietti D, Chemouny JM, Ayari H, Pierre M, et al. Value of biomarkers for predicting immunoglobulin A vasculitis nephritis outcome in an adult prospective cohort. Nephrol Dial Transplant 2018;33(9): 1579-90.
6. Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein Purpura in adults: Outcome and prognostic factors. J Am Soc Nephrol 2002;13:1271-8.
7. Watts RA, Lane S, Scott DGI. What is known about the epidemiology of the vasculitides? Best Pract Res Clin Rheumatol 2005;19:191-207.
8. Maisons V, Ramdani Y, Messiaen C, Sautenet B, Halimi JM, Maillot F, et al. Épidémiologie de la vascularite à IgA de l’enfant et de l’adulte en France à partir de la BNDMR. Rev Medecine Interne 2022;43:A380.
9. Nikolaishvili M, Pazhava A, Di Lernia V. Viral infections may be associated with Henoch–Schönlein Purpura. J Clin Med 2023;12:697.
10. Ramdani Y, Galempoix JM, Augusto JF, Dekmeer E, Perard L, Ferreira N, et al. Immunoglobulin a vasculitis following COVID-19: A French multicenter case series. J Rheumatol 2022;49:1390-4.
11. Batu ED, Sarı A, Erden A, Sönmez HE, Armağan B, Kalyoncu U, et al. Comparing immunoglobulin A vasculitis (Henoch-Schönlein purpura) in children and adults: A single-centre study from Turkey. Scand J Rheumatol 2018;47:481-6.
12. Ramdani Y, Bettuzzi T, Bouznad A, Delaitre L, Nassarmadji K, Didier K, et al. IgA vasculitis following COVID-19 vaccination: A French multicentre case series including 12 patients. J Rheumatol 2023;50(2):252-7.
13. Hankard A, Michot JM, Terrier B, Brihaye B, Chanal J, Combe C, et al. New insights on IgA vasculitis with underlying solid tumor: A nationwide French study of 30 patients. Clin Rheumatol 2021;40:1933-40.
14. Umemura H, Yamasaki O, Iwatsuki K. Leukocytoclastic vasculitis associated with immunoglobulin A lambda monoclonal gammopathy of undetermined significance: A case report and review of previously reported cases. J Dermatol 2018;45:1009-12.
15. Elhani I, Pillebout E, Terrier B, Hankard A, Vrtovsnik F, Jourde-Chiche N, et al. IgA vasculitis with underlying liver cirrhosis: A French nationwide case series of 20 patients. J Rheumatol 2021;48:735.
16. Hočevar A, Rotar Z, Jurčić V, Pižem J, Čučnik S, Vizjak A, et al. IgA vasculitis in adults: The performance of the EULAR/PRINTO/PRES classification criteria in adults. Arthritis Res Ther 2016;18:58.
17. Audemard-Verger A, Pillebout E, Amoura Z, Cacoub P, Jourde-Chiche N, Lioger B, et al. Gastrointestinal involvement in adult IgA vasculitis (Henoch-Schönlein purpura): Updated picture from a French multicentre and retrospective series of 260 cases. Rheumatol Oxf Engl 2020;59:3050-7.
Dans cet article

Résumé
La vascularite à IgA, anciennement appelée purpura rhumatoïde, est une vascularite systémique liée à la présence de dépôts d’immunoglobulines A dans les vaisseaux de petit calibre. Elle fait souvent suite à un facteur déclenchant, comme une infection du tractus respiratoire, la prise d’un médicament ou une vaccination. Elle est caractérisée par la présence d’un purpura vasculaire associé à une atteinte articulaire (arthralgies typiquement des chevilles), gastro-intestinale (douleurs abdominales) et parfois rénale (glomérulonéphrite). Le pronostic vital peut être engagé par l’atteinte gastro-intestinale (risque de perforation digestive), alors que le pronostic à long terme est lié à l’atteinte rénale (risque d’insuffisance rénale chronique). La maladie évolue généralement favorablement, et seul un traitement symptomatique est conseillé. Dans les formes plus sévères, des corticostéroïdes, associés à un immunosuppresseur ou une biothérapie, peuvent être discutés au cas par cas.