Pathologiques : connaître les principaux types de vascularite systémique, les organes cibles, les outils diagnostiques et les moyens thérapeutiques.
Définition et histologie
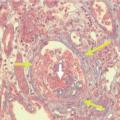
Une vascularite est définie par une inflammation de la paroi vasculaire par un infiltrat leucocytaire fait de lymphocytes et de polynucléaires neutrophiles (PNN) en proportions variables. La destruction rapide des PNN, avec fragmentation nucléaire, est désignée sous le terme de leucocytoclasie. L’infiltrat peut être également constitué d’autres cellules, différant selon la vascularite : polynucléaires éosinophiles dans la granulomatose à éosinophiles avec polyangéite (GEPA), macrophages avec cellules géantes dans les vascularites granulomateuses (maladie de Horton ou artérite à cellules géantes, granulomatose avec polyangéite [GPA], GEPA).
La présence de nécrose fibrinoïde (modification de la substance fondamentale au contact de l’infiltrat de polynucléaires) au sein de la paroi vasculaire caractérise les vascularites nécrosantes (périartérite noueuse [PAN], GPA, GEPA).
L’infiltrat inflammatoire de la paroi vasculaire des petits vaisseaux favorise l’extravasation des hématies, à l’origine du purpura. L’évolution vers la fibrose et la sclérose peut entraîner des sténoses principalement dans les vascularites des gros vaisseaux (maladie de Takayasu). Les lésions endothéliales favorisent les thromboses, et la destruction de la média, les anévrismes (maladie de Behçet).
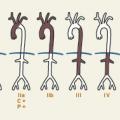
Les premières classifications (anatomopathologiques) distinguaient les vascularites selon l’aspect histologique (
Le diagnostic de vascularite est évoqué devant des manifestations cliniques, ou biologiques. Une preuve histologique obtenue par biopsie d’un tissu accessible (peau, muscle, nerf, rein) est souvent nécessaire.
Des critères de classification des maladies systémiques, et notamment des vascularites, ont été établis, notamment par l’American College of Rheumatology (ACR) et par l’European Alliance of Associations for Rheumatology (EULAR), d’après l’analyse de cohortes de malades (
Classification des vascularites
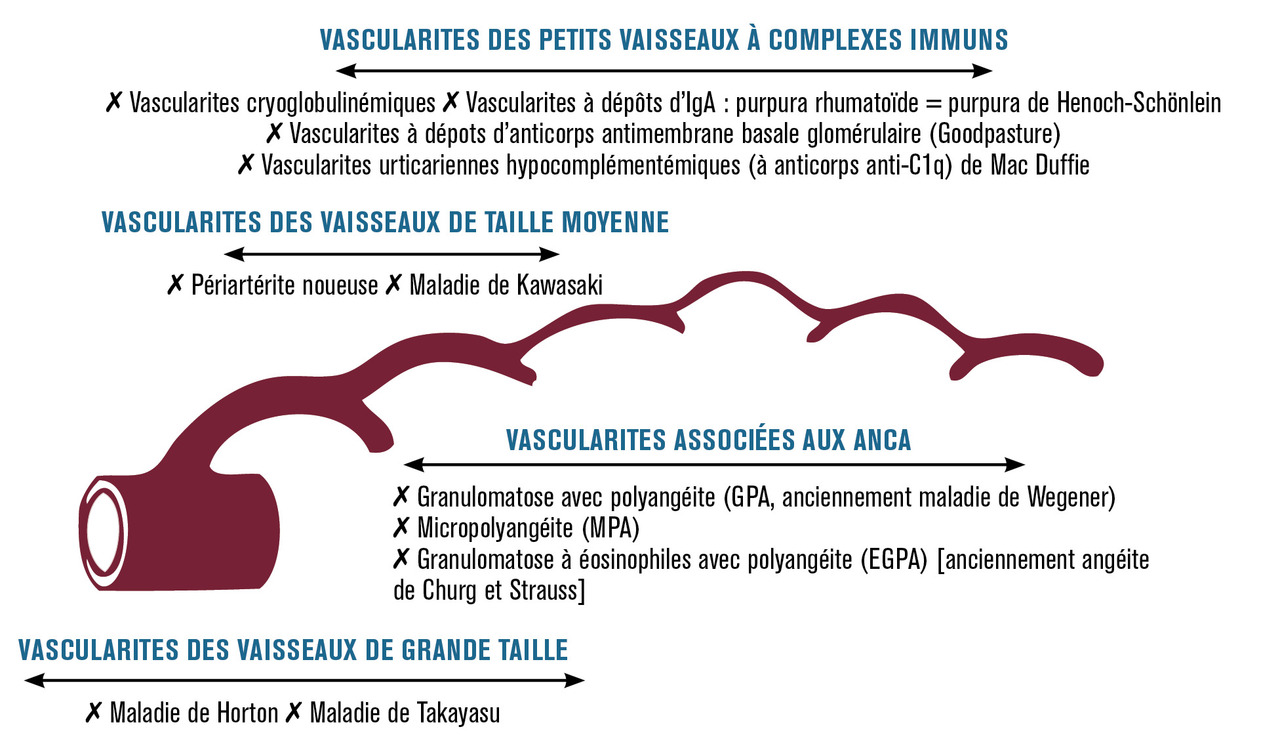
Les manifestations cliniques des vascularites sont fonction du type et de la taille des vaisseaux atteints et de la présence de certains anticorps (anticorps anticytoplasme des polynucléaires, ou antineutrophil cytoplasmic antibodies [ANCA]…) ou de dépôts d’immunoglobulines (immunoglobulines A [IgA], cryoglobulines…).
La classification actuelle (consensus international de Chapel Hill de 2012) prend en compte ces éléments mais reste forcément schématique (
Certaines vascularites touchant préférentiellement une taille de vaisseau peuvent, de façon plus rare, atteindre un vaisseau de taille différente, habituellement plus petit (la maladie de Horton peut atteindre des artères rétiniennes, la périartérite noueuse les petits vaisseaux…).
Vascularites des vaisseaux de gros calibre
Elles concernent l’aorte et ses principales branches.
Artérite à cellules géantes (maladie de Horton)
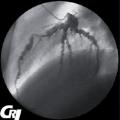
C’est une vascularite granulomateuse survenant préférentiellement après 60 ans. L’atteinte de la carotide externe et de ses branches, typiquement les artères temporales, expliquent la symptomatologie (céphalées, claudication de la mâchoire…).
Les autres vaisseaux peuvent être atteints : aorte, carotide interne, artères vertébrales et sous-clavières. Elle peut être associée à la pseudopolyarthrite rhizomélique. La cécité par névrite optique ischémique antérieure aiguë constitue sa principale complication. Le diagnostic est confirmé par biopsie de l’artère temporale. La corticothérapie est rapidement efficace (
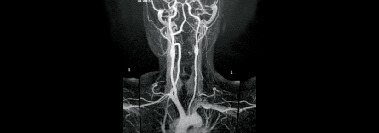
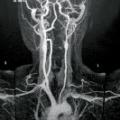
Maladie de Takayasu
Vascularite granulomateuse de l’aorte et de ses principales branches également (
Vascularites des vaisseaux de moyen calibre
Elles concernent les principales artères et veines viscérales et leurs branches initiales.
Périartérite noueuse
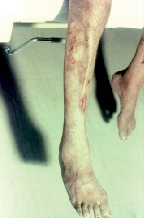
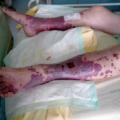
C’est une vascularite nécrosante des vaisseaux de moyen et petit calibres touchant préférentiellement l’adulte entre 40 et 60 ans. Elle est à l’origine de signes cutanés (purpura, nodules, livedo, ulcérations). Des signes généraux (amaigrissement, fièvre, altération de l’état général), une atteinte neurologique périphérique à type de mono- ou multinévrite (sciatique poplitée externe [SPE] [
Le diagnostic est histologique : biopsie cutanée, musculaire, neuromusculaire. La biopsie rénale n’a pas d’intérêt ; elle est même contre-indiquée du fait de l’existence potentielle de microanévrismes. En cas de doute diagnostique, elle ne peut être effectuée qu’après réalisation d’une imagerie artérielle (angioscanner, angio-IRM). La recherche d’ANCA est négative. L’association classique à une infection par le virus de l’hépatite B est désormais exceptionnelle (
Maladie de Kawasaki
La maladie de Kawasaki est une vascularite des vaisseaux de moyen et petit calibres affectant le nourrisson et le jeune enfant de 3 mois à 5 ans.
Les signes cliniques (syndrome adéno-cutanéo-muqueux fébrile) associent :
- une fièvre prolongée inexpliquée (durant plus de 5 jours) ne répondant ni aux antipyrétiques ni aux antibiotiques ;
- une éruption cutanée maculopapuleuse avec érythème palmoplantaire évoluant vers une desquamation ;
- une conjonctivite ;
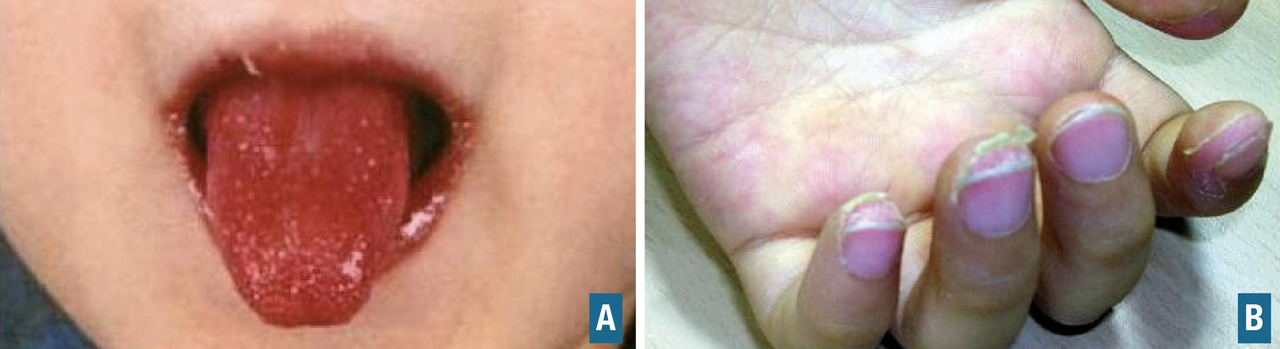
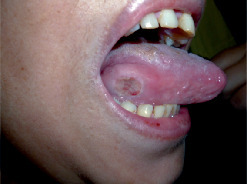
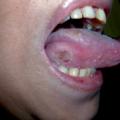
- une atteinte oropharyngée (érythème des lèvres, sécheresse, fissures, langue framboisée [
fig. 6 ]) ; - des adénopathies cervicales (
tableau 5 ).
L’absence de traitement expose au risque de survenue d’anévrismes coronariens (
Vascularites des vaisseaux de petit calibre
Elles concernent les artères intraparenchymateuses, artérioles, capillaires, veinules et veines.
Toutes les vascularites des petits vaisseaux peuvent entraîner :
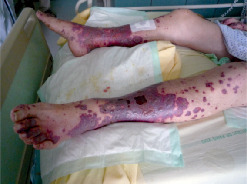
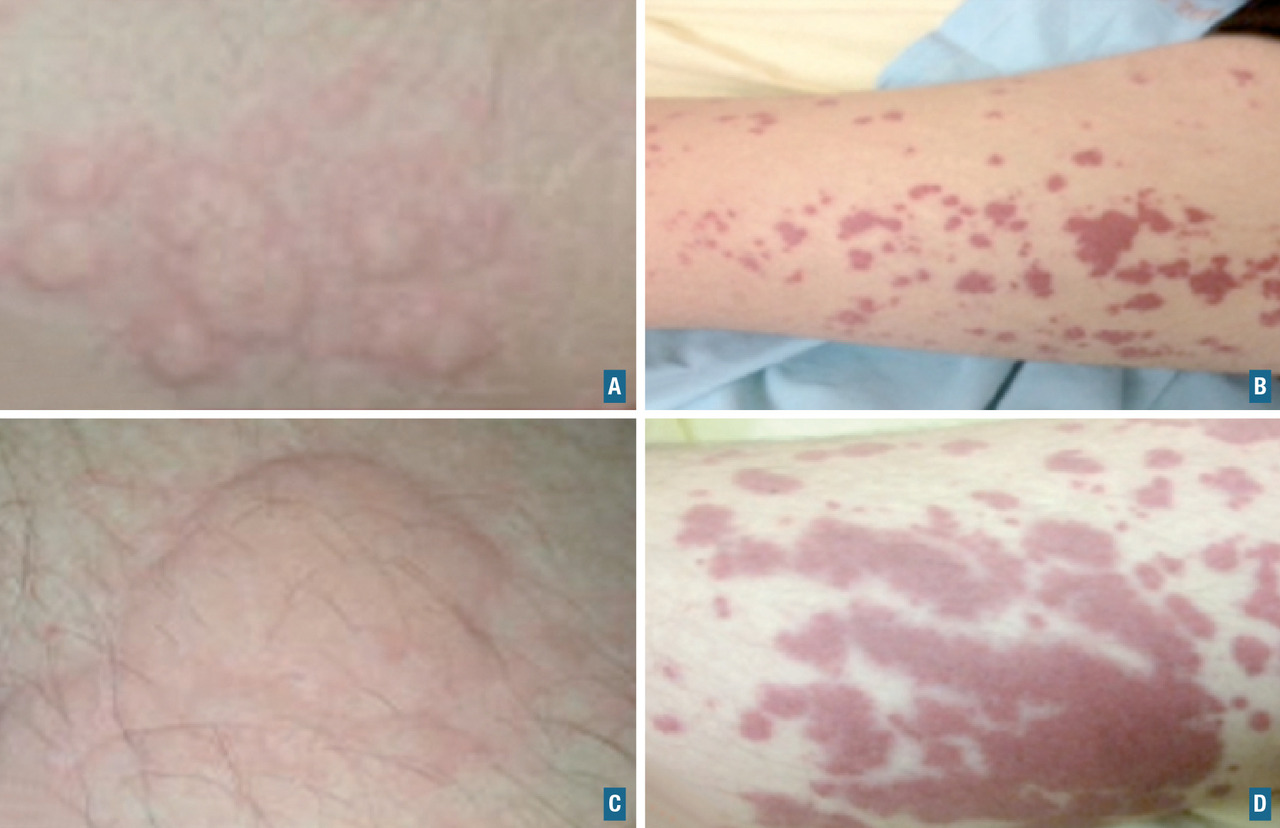
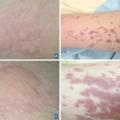
- des manifestations cutanées : purpura, nodules, livedos, ulcérations, nécroses. Le purpura est un purpura vasculaire, prédominant au niveau des membres inférieurs, le plus souvent infiltré et constitué d’éléments différents (urticaire, bulles ou zones nécrotiques) ;
- des manifestations articulaires : arthralgies, polyarthralgies, polyarthrite ;
- des manifestations systémiques : fièvre, altération de l’état général, amaigrissement ;
- un syndrome inflammatoire biologique (vitesse de sédimentation [VS] et protéine C réactive [CRP] élevées).
Les autres manifestations cliniques et biologiques varient selon le type de vascularite.
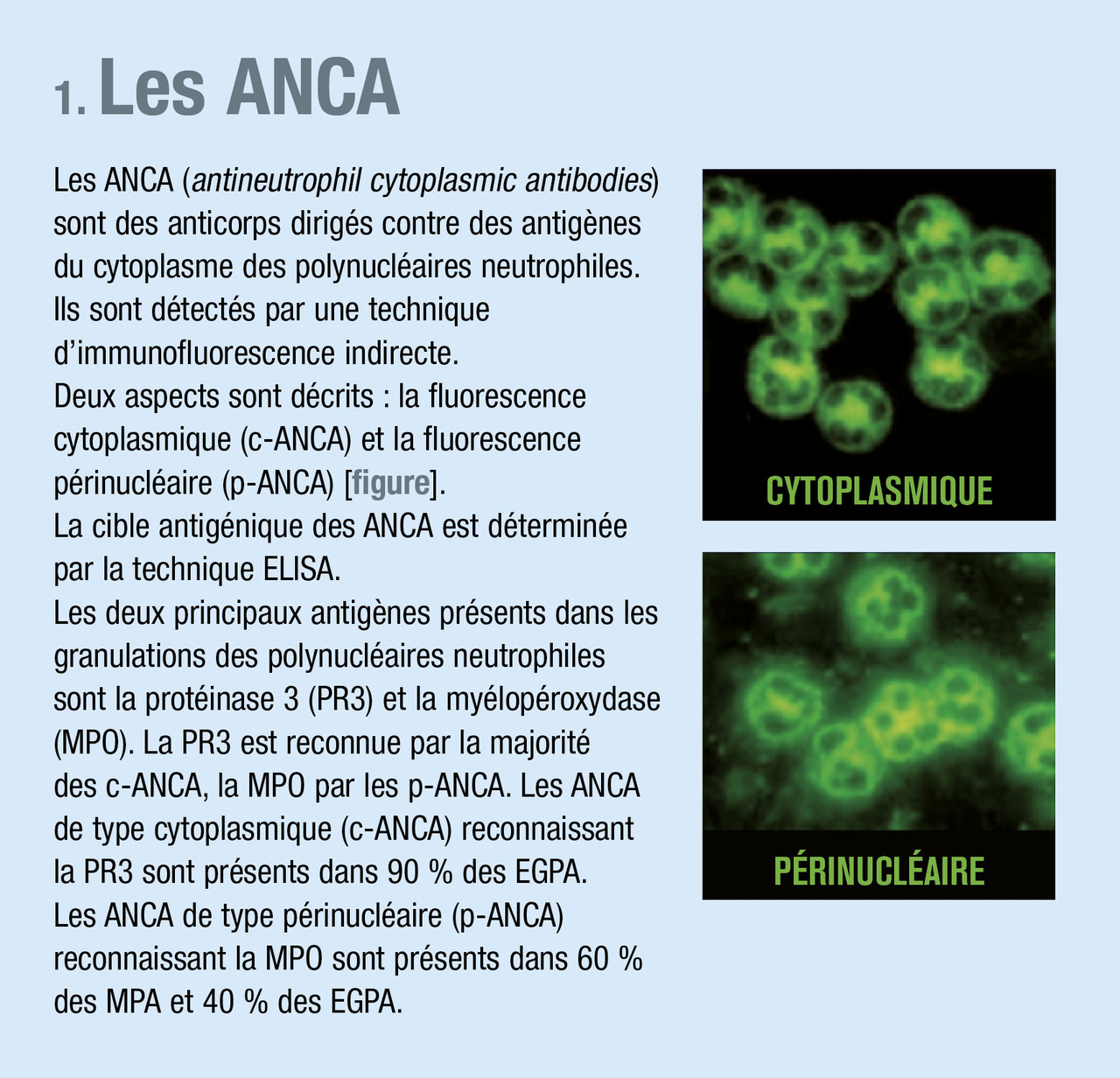
Vascularites associées aux ANCA (anticorps anticytoplasme des polynucléaires)
Ce sont des vascularites nécrosantes des petits vaisseaux caractérisées par l’absence ou la rareté de dépôts immuns dans la paroi des vaisseaux, (ce qui les différencie des vascularites des petits vaisseaux à complexes immuns [vascularites cryoglobulinémiques…]), mais par la présence dans le sérum d’anticorps anticytoplasme des polynucléaires (
Granulomatose avec polyangéite
Associant des lésions histologiques de vascularite et de granulomatose, elle se caractérise par trois atteintes préférentielles (ORL, pulmonaire et rénale [glomérulaire]) et par la présence d’ANCA de fluorescence cytoplasmique (immunofluorescence indirecte) à spécificité protéinase 3 (c-ANCA PR3) [ELISA, enzyme-linked immunosorbent assay] dans plus de 90 % des cas (
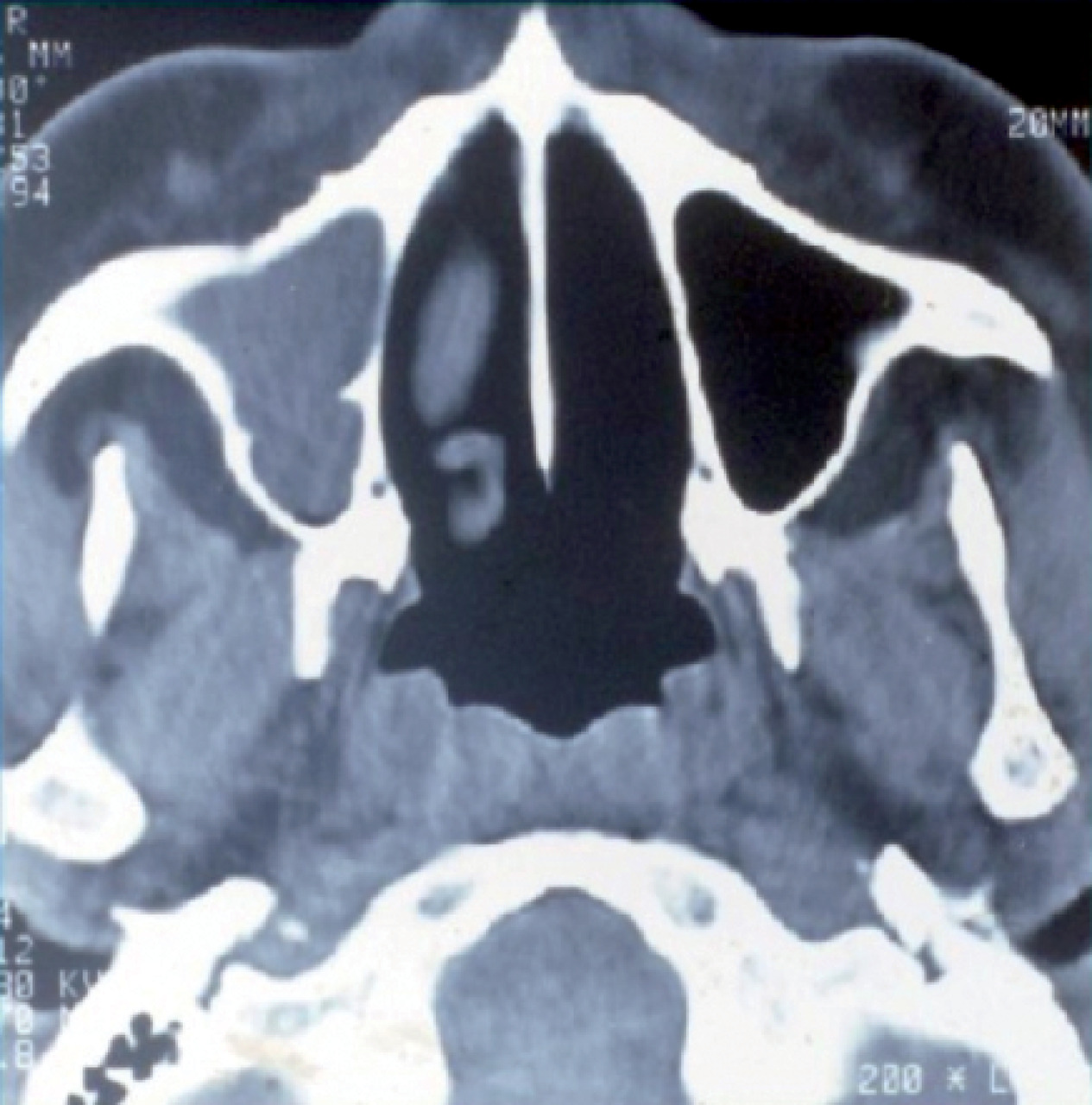
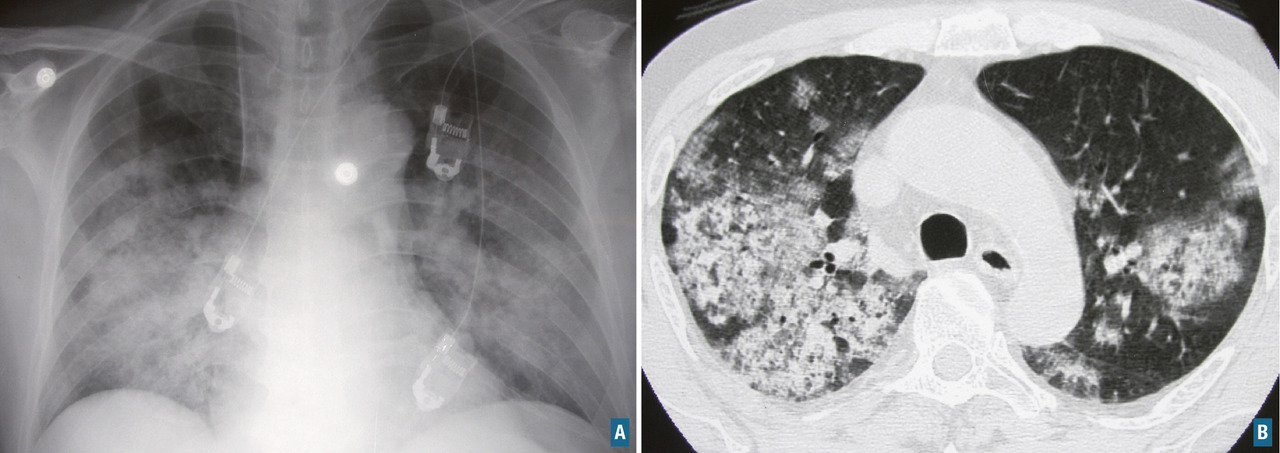
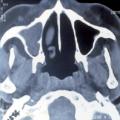
L’atteinte ORL se traduit par des manifestations initialement non spécifiques : rhinorrhée, épistaxis, sinusite ou otite séreuse, pouvant persister durant plusieurs semaines ou mois. La tomodensitométrie montre un comblement des sinus, qui peut évoluer vers une destruction osseuse (
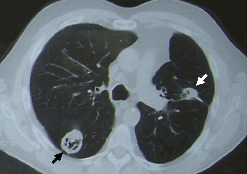
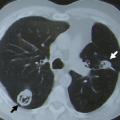
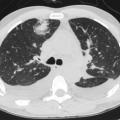
L’atteinte pulmonaire est variable : infiltrats alvéolaires, aspect en verre dépoli, nodules multiples excavés (
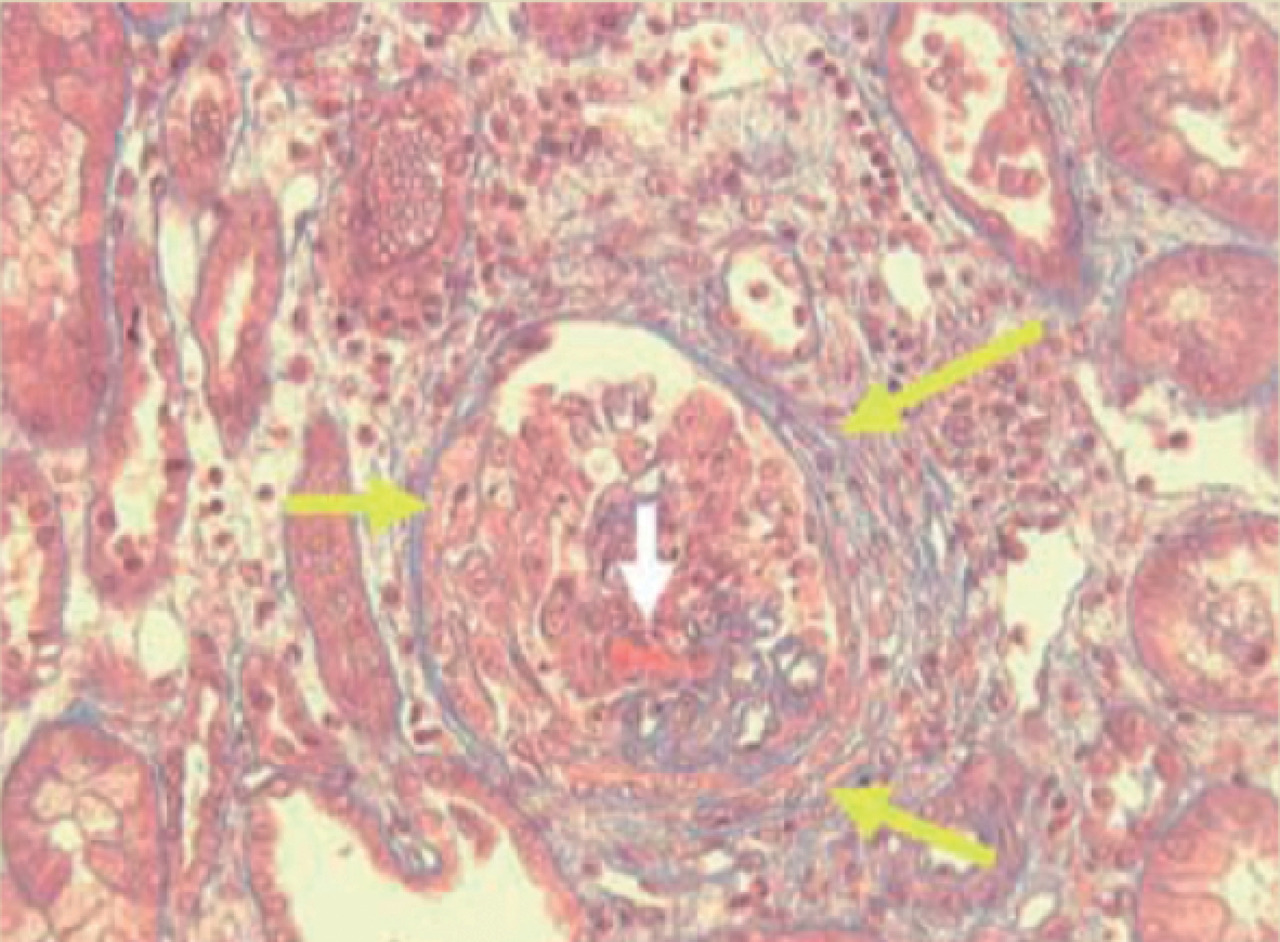
L’atteinte rénale est glomérulaire pauci-immune (absence de dépôts significatifs d’immunoglobulines ou de complément dans les glomérules), nécrosante, segmentaire et focale, associée à une prolifération extracapillaire (
Micropolyangéite ou polyangéite microscopique
C’est une vascularite des petits vaisseaux (artérioles, capillaires et veinules) se manifestant par une glomérulonéphrite pauci-immune, nécrosante, segmentaire et focale et associée à une prolifération extracapillaire identique à celle de la granulomatose avec polyangéite, si ce n’est l’absence de granulomes. L’atteinte pulmonaire est présente dans 20 à 30 % des cas sous forme d’hémorragies alvéolaires. D’autres atteintes sont possibles : cutanées, articulaires, digestives. Les ANCA, présents dans 60 % des cas, sont de fluorescence périnucléaire et de spécificité myéloperoxydase (p-ANCA MPO) [ELISA].
Granulomatose éosinophile avec polyangéite
La granulomatose éosinophile avec polyangéite (GEPA)est une vascularite systémique et pulmonaire des artères et des veines de petit calibre. Les lésions histologiques associent une nécrose fibrinoïde de la média, des infiltrats tissulaires à éosinophiles et des granulomes extravasculaires. Elle survient préférentiellement sur terrain atopique et chez les asthmatiques (
L’évolution se fait classiquement en trois phases :
- l’apparition d’un asthme (tardif, après 30 ans) rapidement corticodépendant et de manifestations ORL (rhinite, sinusite) ;
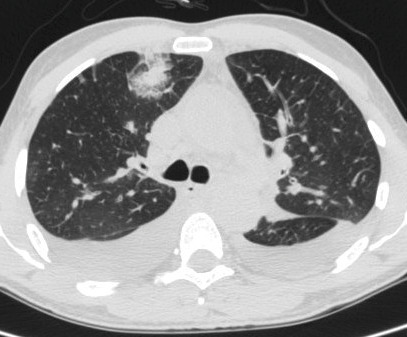
- puis une hyperéosinophilie sanguine et tissulaire, à l’origine d’opacités alvéolaires localisées non systématisées périphériques, uni- ou bilatérales, généralement à type d’infiltrats (
fig. 12 ) ;
- enfin une vascularite systémique avec altération de l’état général, manifestations neurologiques (multinévrite), cutanées (purpura, livedo…), digestives (douleurs abdominales, diarrhée…), rénales (glomérulonéphrites extracapillaires). L’atteinte cardiaque (péricardique ou myocardique), présente dans 35 % des cas, est la première cause de mortalité spécifique. L’atteinte myocardique est plus fréquente chez les patients n’ayant pas d’ANCA.
L’hyperéosinophilie sanguine (importante > 1 500/mm3) et l’élévation des immunoglobulines E (IgE) sériques (non spécifique) sont constantes.
Les ANCA ne sont présents que dans 40 % des cas et sont de fluorescence périnucléaire (en immunofluorescence) et de spécificité MPO (ELISA).
Évoqué sur l’association des manifestations cliniques (asthme…) et biologiques (hyperéosinophilie, ANCA…), le diagnostic peut nécessiter, en cas de doute, une confirmation histologique par une biopsie cutanée, neurologique ou musculaire.
Vascularites à complexes immuns
Vascularites à IgA (purpura rhumatoïde)
La vascularite à IgA est une vascularite des petits vaisseaux liée à des dépôts d’immunoglobulines A. Elle est plus fréquente chez l’enfant âgé de 3 et 15 ans mais possible chez l’adulte et, dans ce cas, plus sévère. La triade caractéristique associe un purpura vasculaire (
Le traitement comporte un repos au lit, qui permet de diminuer les poussées cutanées mais n’influe pas sur la durée ou l’évolution de la maladie. Une corticothérapie est indiquée dans les formes sévères (atteinte digestive non soulagée par le traitement symptomatique, atteinte rénale après biopsie).
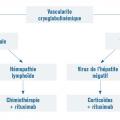
Vascularites cryoglobulinémiques
Les cryoglobulines sont des immunoglobulines présentes dans le sérum, précipitant à des températures inférieures à 37 °C et se dissolvant lors du réchauffement.
Trois types en sont décrits :
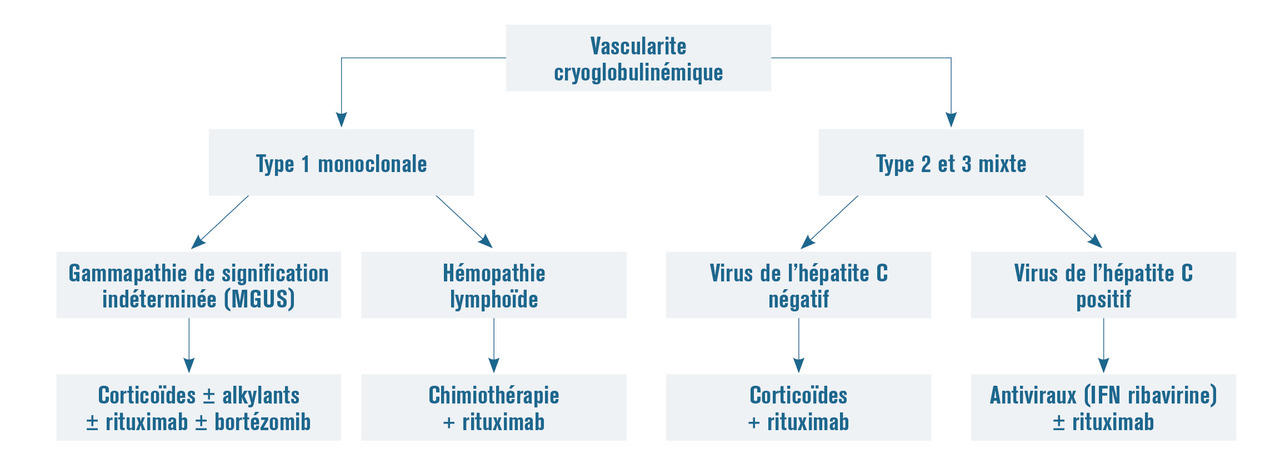
- les cryoglobulinémies de type 1 sont monoclonales, plus souvent IgM et sont associées à une hémopathie lymphoïde (leucémie lymphoïde chronique, maladie de Waldenström…), un myélome ou une gammapathie monoclonale de signification indéterminée (MGUS) ;
- les cryoglobulinémies de types 2 et 3 sont mixtes car composées d’immunoglobulines polyclonales associées à une immunoglobuline monoclonale (type 2) ou non (type 3). Les cryoglobulinémies de type 2 sont composées d’immunoglobulines de classes différentes dont l’une est monoclonale de type IgM et dirigée contre une IgG polyclonale. Celles de type 3 correspondent à l’association d’immunoglobulines de classes différentes IgG et IgM polyclonales. La majorité des cryoglobulinémies mixtes (types 2 et 3) s’observe au cours de l’infection par le virus de l’hépatite C (VHC) et plus rarement dans les maladies auto-immunes et certaines hémopathies lymphoïdes B.
Les cryoglobulines sont à l’origine d’une vascularite des petits vaisseaux avec dépôts hyalins intravasculaires, correspondant en immunofluorescence au cryoprécipité constitué par les immunoglobulines.
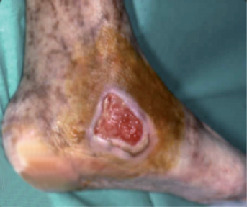
Les manifestations cliniques associent un purpura vasculaire, des ulcérations cutanées (
Le diagnostic est confirmé par la découverte de la cryoglobulinémie. Sa recherche nécessite des conditions de recueil adaptées : prélèvement et acheminement au laboratoire à 37 °C afin d’éviter une précipitation dans le tube. La présence d’une cryoglobuline se traduit souvent par une baisse du complément (CH50 et fraction C4) et la positivité du facteur rhumatoïde.
Le traitement des vascularites cryoglobulinémiques dépend du type de cryoglobulinémie. Les cryoglobulinémies de type 1 associées à une gammapathie de signification indéterminée (MGUS) sont traitées comme le myélome, celles associées à une hémopathie lymphoïde par chimiothérapie et rituximab. La prise en charge des cryoglobulinémies mixtes consiste initialement en un traitement antiviral pour les formes associées au virus de l’hépatite C, complété par le rituximab. Les formes non associées au VHC doivent être traitées par rituximab (fig. 15).
Vascularites à dépôts d’anticorps anti-membrane basale glomérulaire (syndrome de Goodpasture)
Cette vascularite rare des petits vaisseaux atteint l’homme entre 30 et 45 ans. Elle est responsable d’un syndrome pneumorénal superposable à celui des vascularites à ANCA (granulomatose avec polyangéite et micropolyangéite). L’insuffisance rénale aiguë par glomérulonéphrite rapidement progressive est associée à une détresse respiratoire par hémorragie alvéolaire. Le diagnostic est confirmé par la présence d’anticorps antimembrane basale glomérulaire circulants et de dépôts linéaires d’IgG le long de la membrane basale glomérulaire sur la biopsie rénale, qui objective une glomérulonéphrite à croissants extracapillaires. Le traitement associe les corticoïdes, les échanges plasmatiques et le cyclophosphamide.
Vascularites uticariennes hypocomplémentémiques
Elles sont définies par des poussées d’urticaire fixes (plus de 24 heures) sur une période de plus de six mois, une vascularite leucocytoclasique (purpura vasculaire) (fig. 16) et une hypocomplémentémie (baisse des fractions C4 et C1q). Des manifestations systémiques peuvent s’y associer : arthralgies, angioœdèmes, douleurs abdominales, uvéites, épisclérites, glomérulonéphrites. Une bronchopneumopathie chronique obstructive, parfois au stade d’insuffisance respiratoire chronique, des péricardites, des valvulopathies et des atteintes neurologiques ont également été rapportées. Les anticorps anti-C1q sont le plus souvent présents. Cette vascularite précède ou s’associe au lupus érythémateux systémique dans plus de 50 % des cas. Le traitement des formes cutanées isolées repose sur la dapsone, l’hydroxychloroquine ou la colchicine. En cas d’atteinte viscérale, les corticoïdes et les immunosuppresseurs sont nécessaires.
Autres vascularites
Vascularites touchant des vaisseaux de différentes tailles et de différents types
Elles peuvent concerner les vaisseaux de petit, moyen et gros calibres, et tant les artères que les veines et les capillaires.
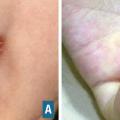
Maladie de Behçet
La maladie de Behçet a une distribution géographique particulière (bassin méditerranéen, Japon). L’existence d’une aphtose en est caractéristique si elle est bipolaire (buccale et génitale) (fig. 17). La notion de prédisposition génétique est confortée par la présence (inconstante) du phénotype HLA-B51. Les autres manifestations cutanées sont l’érythème noueux et la pseudofolliculite. L’atteinte ophtalmologique est fréquente, sous la forme d’une uvéite antérieure, postérieure (avec vascularite rétinienne) ou totale (panuvéite). D’autres complications graves sont possibles :
- atteinte vasculaire veineuse : thromboses veineuses superficielles ou profondes (veine cave, veines sus-hépatiques, veines cérébrales…) ;
- atteinte vasculaire artérielle (thromboses et anévrismes ; artères pulmonaires, aorte) ;
- atteinte neurologique centrale multifocale (méningite, encéphalite, myélite, thrombose veineuse centrale) [
tableau 9 ].
Le traitement des aphtes est fondé sur la colchicine. Les autres atteintes sont traitées en fonction de leur gravité par une corticothérapie orale, des immunosuppresseurs, des anti-TNF.
Syndrome de Cogan
Le syndrome de Cogan, affection très rare, est caractérisé par une atteinte ophtalmologique (kératite interstitielle) et audiovestibulaire (syndrome vestibulaire avec vertiges, acouphènes) évoluant rapidement vers la surdité. Une vascularite des gros vaisseaux peut être associée (insuffisance aortique par aortite), de même qu’une vascularite des vaisseaux de taille moyenne. Le traitement est fondé sur la corticothérapie.
Vascularites touchant un seul organe (artère ou veine quelle que soit leur taille)
Elles peuvent rester isolées ou s’intégrer ultérieurement dans le cadre d’une vascularite systémique.
Vascularites cutanées leucocytoclasiques
Elles sont définies par un infiltrat à polynucléaires neutrophiles dont le noyau est fragmenté. Elles se traduisent par un purpura vasculaire, des lésions urticariennes, voire nécrotiques. Elles peuvent être d’origine infectieuse (endocardites, méningococcémies, rickettsioses…), médicamenteuses ou liées à des maladies systémiques, des cancers, des hémopathies…
Vascularites du système nerveux central
Toute vascularite systémique peut donner une vascularite du système nerveux central, mais certaines vascularites n’entraînent qu’une atteinte isolée, touchant de façon prédominante les artères de petit calibre et responsable de manifestations neurologiques centrales polymorphes (céphalées, encéphalopathie et déficits focaux...).
Aortites
Elles sont décrites dans de nombreuses pathologies (syphilis, rickettsioses, sarcoïdose, syndrome de Cogan…) Les aortites isolées peuvent être considérées comme des équivalents de la maladie de Takayasu.
Vascularites secondaires
Les vascularites peuvent compliquer d’autres maladies systémiques : lupus, polyarthrite rhumatoïde, sarcoïdose, ou être associées à une cause différente.
Elles s’intègrent alors dans les catégories précédentes selon leurs caractéristiques : vascularites cryoglobulinémiques associées au virus de l’hépatite C, vascularites associées au virus de l’hépatite B, aortite syphilitique, vascularites médicamenteuses à complexes immuns, vascularites médicamenteuses associées aux ANCA, vascularites associées aux cancers (et hémopathies).
Les vascularites médicamenteuses sont essentiellement responsables de manifestations cutanées à type de purpura, mais pas d’atteinte systémique. Les médicaments le plus souvent en cause sont les sulfamides antibactériens et antidiabétiques, les bêtalactamines, les tétracyclines, les diurétiques thiazidiques, l’allopurinol, les anti-inflammatoires non stéroïdiens. Les antithyroïdiens de synthèse (méthimazole, propylthio-uracile, benzythio-uracile) peuvent être à l’origine de vascularites systémiques avec présence d’ANCA.
Autre entité proche des vascularites : la maladie de Buerger
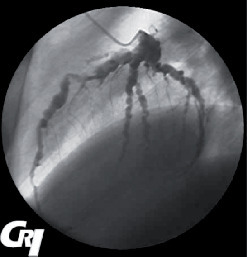
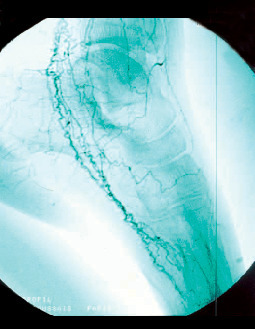
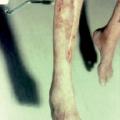
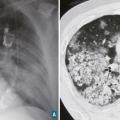
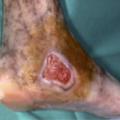
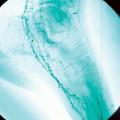
La maladie de Buerger ou thromboangéite oblitérante est une artériopathie non athéroscléreuse, segmentaire et occlusive touchant principalement le sujet jeune de sexe masculin et fumeur. Elle est caractérisée par la présence de thrombus occlusifs, inflammatoires des artères de petit et moyen calibre et des veines des extrémités des membres, mais l’infiltrat inflammatoire de la paroi est inconstant. Les signes cliniques sont un syndrome de Raynaud, une artériopathie digitale, des thromboses superficielles migratrices (
L’imagerie met en évidence une atteinte occlusive segmentaire des artères de petit et moyen calibres, initialement des artères digitales. La présence d’une circulation collatérale est fréquente, entraînant un aspect tortueux avec des images en « tire-bouchon » (fig. 18). Les artères proximales sont normales. L’arrêt du tabac est indispensable. L’iloprost (analogue des prostacyclines) améliore les lésions ischémiques distales.
Vous pouvez retrouver un quiz lié à cet item sur notre site internet : https://www.larevuedupraticien.fr/ les-tests/les-quiz
POINTS FORTS À RETENIR
Les manifestations cliniques des vascularites sont fonction du type et de la taille de vaisseaux atteints et de la présence de certains anticorps (ANCA…) ou de dépôts d’immunoglobulines (IgA, cryoglobulines…).
Vascularites
Maladies de Horton : sujet âgé, céphalées, biopsie artère temporale, risque de cécité, corticoïdes.
Périartérite noueuse : adulte 40-60 ans, multinévrite, altération de l’état général, purpura, livedo, microanévrismes, biopsie musculaire ou neuromusculaire : vascularite nécrosante, ANCA négatifs
Vascularites associées aux ANCA
Granulomatose avec polyangéite (GPA) : adulte, atteinte ORL, atteinte pulmonaire, glomérulonéphrite extracapillaire, c-ANCA PR3
Micropolyangéite (MPA) : adulte, glomérulonéphrite extracapillaire, hémorragie alvéolaire, p-ANCA MPO
Granulomatose éosinophile avec polyangéite (EGPA) : adulte, asthme, infiltrats pulmonaires, glomérulonéphrite extracapillaire, hyperéosinophilie, p-ANCA, MPO
Vascularites à complexes immuns
Vascularites cryoglobulinémiques : purpura, neuropathie périphérique, glomérulonéphrite membranoproliférative, baisse du C4, hépatite C
Vascularites cryoglobulinémiques : enfant, purpura, arthralgies, douleurs abdominales, ± glomérulopathie, biopsie cutanée : dépôts IgA, augmentation IgA sériques
Collège français des pathologistes (CoPath). Coordonné par J.-F. Émile, E. Leteurtre et S. Guyétant. Livre de pathologie générale, 3e édition. Enseignement thématique. Biopathologie tissulaire. Elsevier Masson.
 Encadrés
Encadrés