- Un homme de 32 ans consulte pour des dorsalgies chroniques.
- Il évoque une maladie de Bouveret traitée cinq ans plus tôt, qui a permis de déceler une dilatation aortique modérée.
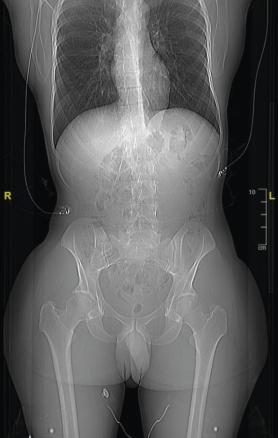
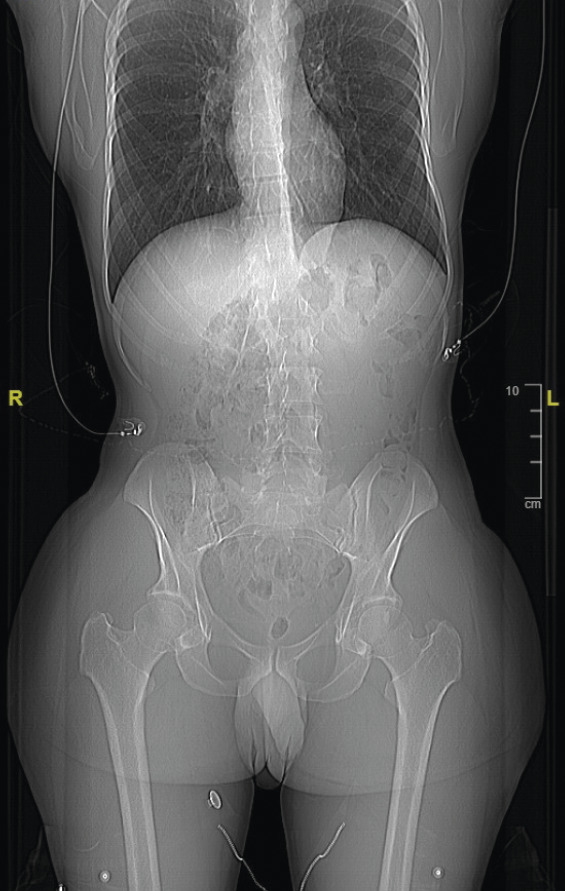
- Il mesure 1,91 m et pèse 92 kg. L’examen révèle une cyphose et une scoliose (
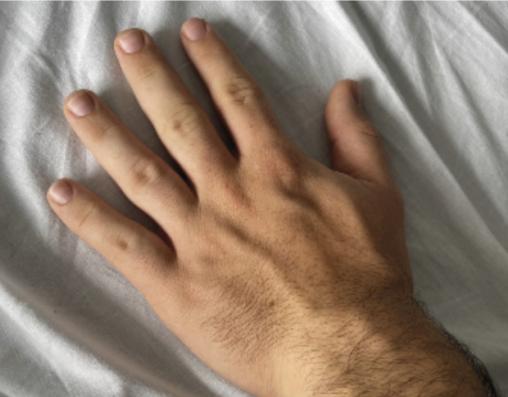
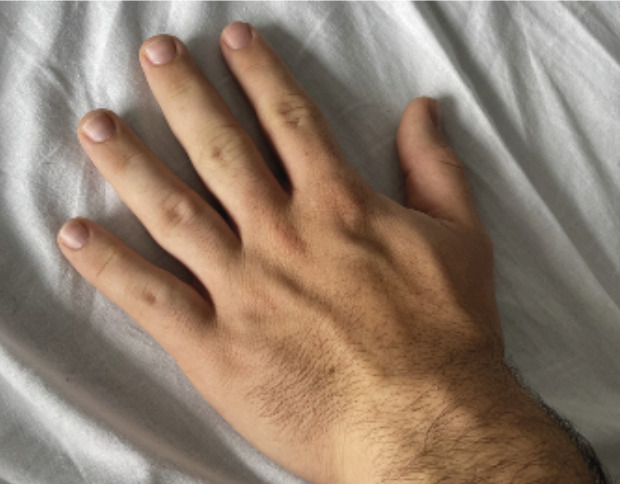
fig. 1 ). Un prognathisme, une macroglossie, une hypertrophie des golfes frontaux, un élargissement des mains (fig. 2 ) et des pieds (il chausse du 47) ainsi que des sueurs nocturnes sont également constatés. - L’association de tous ces signes cliniques fait évoquer le diagnostic d’acromégalie. Le bilan biologique confirme la suspicion clinique avec une IGF-1 (insulin-like growth factor 1) élevée à 721 µg/L (valeurs normales : 88-245 µg/L) et une GH (growth hormone) moyenne à 33,6 µg/L, non freinée par l’hyperglycémie provoquée par voie orale (HGPO).
L’acromégalie est une pathologie rare : son incidence est estimée entre 3 et 15 par million d’habitants par an, et sa prévalence entre 70 et 140 par million d’habitants. Elle est liée à une hypersécrétion de l’hormone de croissance (GH). Sa cause la plus fréquente est l’adénome hypophysaire.
Le diagnostic, essentiellement clinique, demeure délicat du fait de l’installation progressive des symptômes. Une errance d’environ cinq ans est généralement constatée entre les premiers symptômes et la confirmation diagnostique.1
Or le diagnostic précoce et le contrôle optimal de la maladie (clinique, hormonal et tumoral) permettent d’améliorer la qualité de vie des patients, leur morbidité et leur mortalité.
Si le syndrome dysmorphique typique oriente facilement vers une acromégalie, toute la difficulté réside dans le fait de l’évoquer devant des formes plus modérées associées à des signes généraux (sueurs, céphalées, asthénie, paresthésies des mains, syndrome du canal carpien, douleurs articulaires), des signes de complication de la maladie (ostéoarticulaires, cardiovasculaires, syndrome d’apnées du sommeil, diabète, goitre, polypes, coliques), ou en cas d’hyperprolactinémie associée.
Le diagnostic biologique est, quant à lui, relativement simple puisqu’il repose en premier lieu sur le dosage de l’IGF-1, marqueur de la sécrétion pulsatile de GH. Une concentration normale en IGF-1 exclut, dans la majorité des cas, une acromégalie. En revanche, un taux élevé doit amener à orienter le patient vers un endocrinologue pour évaluer la nécessité d’un dosage de GH lors d’une HGPO. L’interprétation des résultats doit tenir compte de l’âge, du sexe, de l’indice de masse corporelle et d’une éventuelle contraception œstroprogestative.
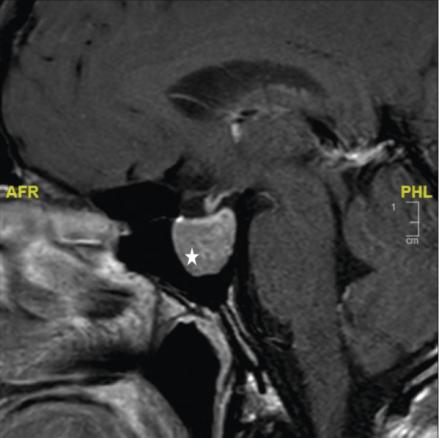
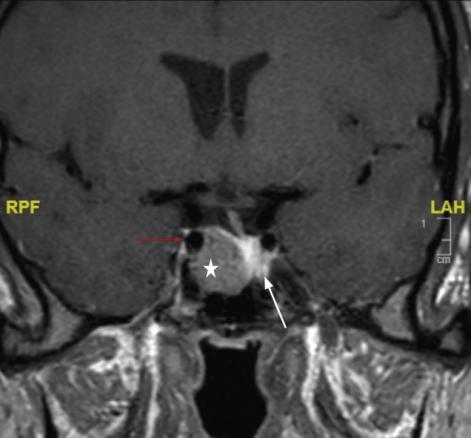
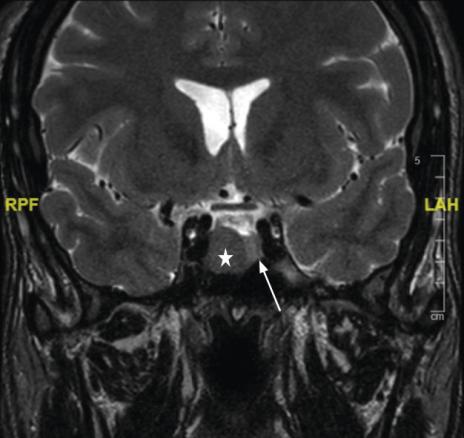
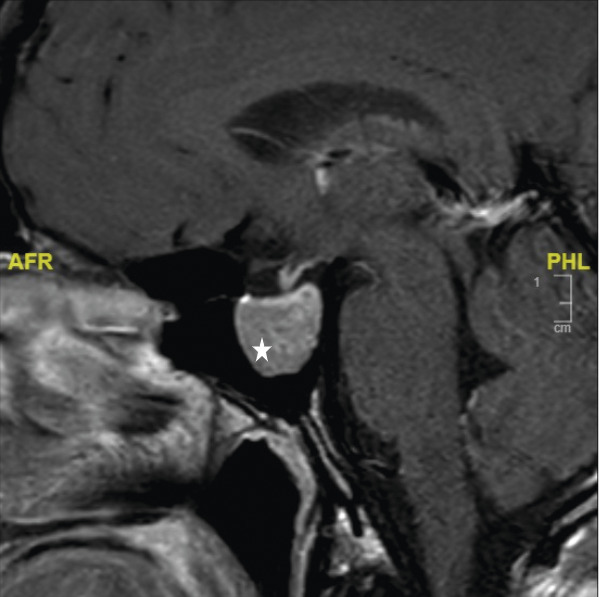
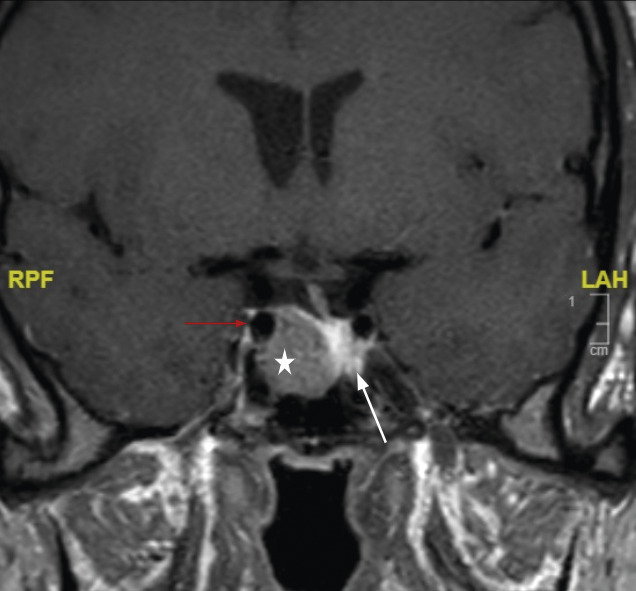
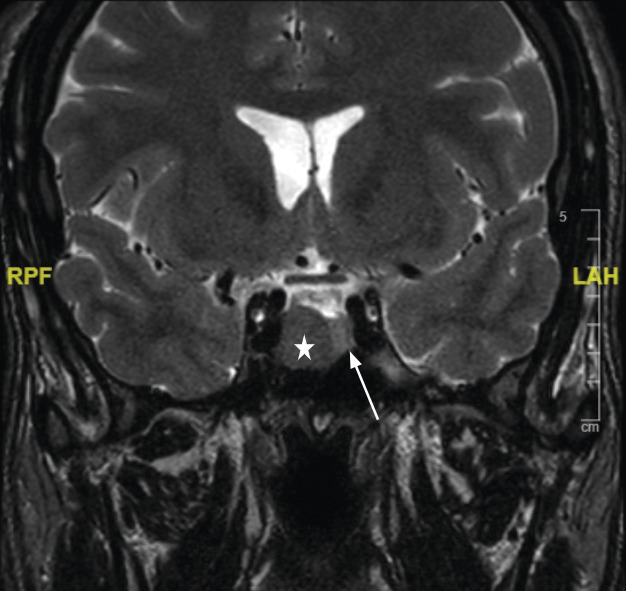
La réalisation d’une IRM hypophysaire permet de visualiser la tumeur (
Le traitement de première intention consiste en une exérèse chirurgicale de la tumeur hypophysaire somatotrope, lorsqu’elle est possible.
Une prise en charge pluridisciplinaire et coordonnée par une équipe d’endocrinologie familière de cette pathologie est indispensable, notamment pour établir la meilleure stratégie thérapeutique. Un protocole national de diagnostic et de soins (PNDS), récemment publié à destination des médecins traitants, précise les modalités de prise en charge diagnostique et thérapeutique optimale ainsi que le parcours de soins, coordonné par le centre de référence des maladies rares de l’hypophyse (CRMR HYPO).2
1. Colao A, Grasso LFS, Giustina A, et al. Acromegaly. Nat Rev Dis Primers 2019;21;5(1):20.
2. CRMR HYPO. Synthèse à destination du médecin traitant. Extraite du Protocole national de diagnostic et de soins (PNDS). Acromégalie. 2021. Disponible sur https://bit.ly/3KRafSv
Une question, un commentaire ?