La pathogenèse de la thrombose associée au cancer est complexe et multifactorielle. Outre les facteurs cliniques généraux (âge, antécédents de maladie thromboembolique veineuse, chirurgie, immobilisation prolongée, etc.), des facteurs spécifiques participent à l’hypercoagulabilité : site du cancer (notamment les tumeurs cérébrales, pancréatiques, gastriques, ovariennes, utérines, pulmonaires et rénales, ainsi que le myélome et les lymphomes hodgkiniens ou non), stade métastatique, thérapies anticancéreuses utilisées et propriétés biologiques prothrombogènes des cellules tumorales. Bien souvent, ces facteurs spécifiques sont également impliqués dans le potentiel agressif des tumeurs.1
Propriétés procoagulantes des cellules cancéreuses
Protéines procoagulantes, microparticules tumorales et protéines de la fibrinolyse sont autant d’acteurs cellulaires ou plasmatiques favorisant la coagulation. Leurs modes d’action sont de mieux en mieux connus.
Protéines procoagulantes
La coagulation in vivo est initiée par l’exposition à la surface cellulaire du facteur tissulaire (FT) qui, complexé au FVII/FVIIa, déclenche le processus en activant les facteurs IX et X. De nombreuses tumeurs solides et hémopathies malignes expriment de manière constitutive le FT, dont le niveau d’expression est corrélé au grade histologique, à la taille de la tumeur et au degré d’angiogenèse.
Certaines cellules tumorales expriment également le facteur « cancer procoagulant » (CP) qui, contrairement au FT, active directement le facteur X, indépendamment du facteur VII. Le CP a été détecté dans différentes cellules malignes, provenant de tumeurs solides et hématologiques ; il n’est pas présent dans les tissus normaux.
Enfin, la quasi-totalité des cellules cancéreuses solides ou hématologiques surexpriment l’héparanase. Cette enzyme interagit avec l’inhibiteur de la voie du facteur tissulaire (TFPI, pour tissue factor pathway inhibitor) à la surface des cellules endothéliales et tumorales, entraînant la dissociation du TFPI et donc une augmentation de l’activité du FT à la surface des cellules.
Microparticules tumorales
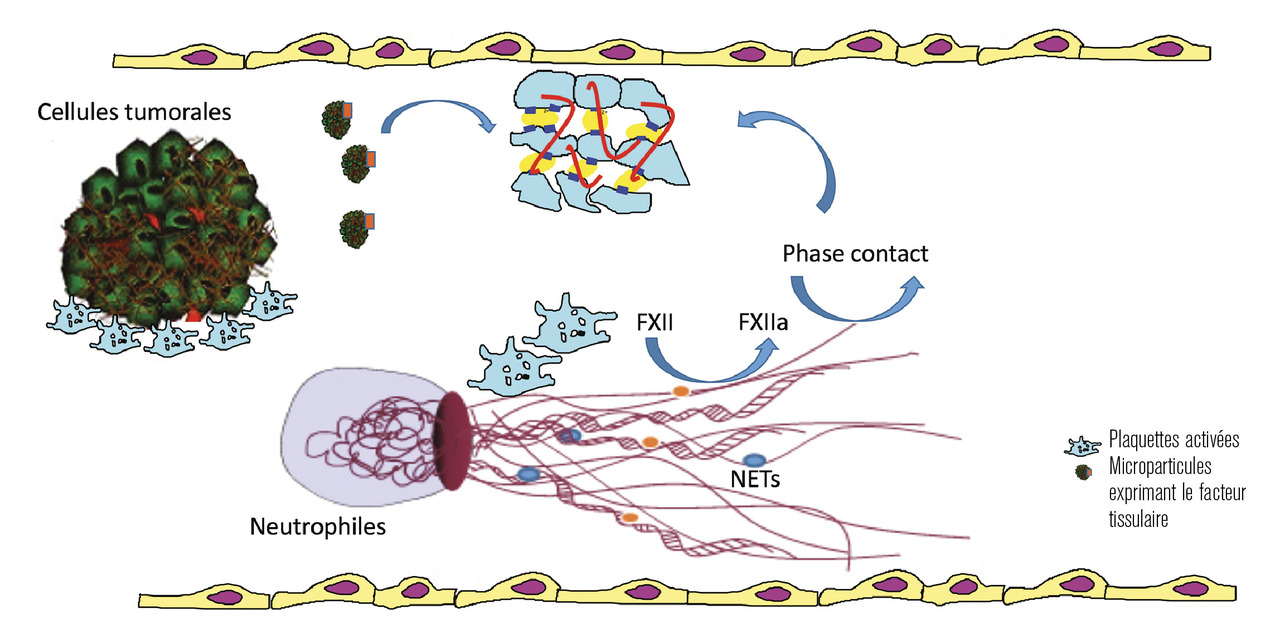
Parallèlement à l’expression cellulaire du FT, il existe un pool circulant plasmatique, essentiellement lié à des microparticules membranaires (MP). Ces dernières sont des vésicules membranaires libérées par presque tous les types de cellules sanguines lors de leur activation, de l’apoptose ou de la transformation maligne.2 Les MP peuvent directement dériver des cellules tumorales. Leur potentiel procoagulant est lié à l’exposition à leur surface de phospholipides chargés négativement (essentiellement phosphatidylsérine) mais également de FT, permettant ainsi l’activation de la coagulation (
Protéines de la fibrinolyse
Les cellules cancéreuses expriment des protéines du système fibrinolytique, notamment les inhibiteurs 1 et 2 de l’activateur du plasminogène (PAI-1 et PAI-2). Des taux élevés de PAI-1 ont été mesurés chez des patients atteints de cancer du pancréas et de gliome malin.
Interactions entre cellules cancéreuses et cellules vasculaires de l’hôte
Les cellules cancéreuses entretiennent des interactions fortes avec les cellules vasculaires normales de l’hôte (en particulier les plaquettes, les leucocytes et les cellules endothéliales) et activent le potentiel procoagulant de ces dernières (
Activation plaquettaire
Les plaquettes jouent un rôle très important dans l’hypercoagulation du cancer, mais également dans le potentiel métastatique des cellules cancéreuses. L’adhésion directe des plaquettes aux cellules cancéreuses « protège » les cellules tumorales de la reconnaissance immunologique des cellules NK. Cette adhésion se fait essentiellement par l’intermédiaire d’un récepteur, la sélectine P. L’activation et l’agrégation plaquettaire sont favorisées par la sécrétion tumorale de molécules activant les plaquettes (adénosine diphosphate [ADP], métalloprotéinases matricielles, interleukine 6 [IL-6]).
Certains adénocarcinomes sécrètent des mucines anormalement glycosylées qui permettent une interaction entre les plaquettes et les leucocytes, médiée par la sélectine P sur les plaquettes et le P-selectin glycoprotein ligand (PSLGI) sur les leucocytes, à l’origine de la formation de thrombus.
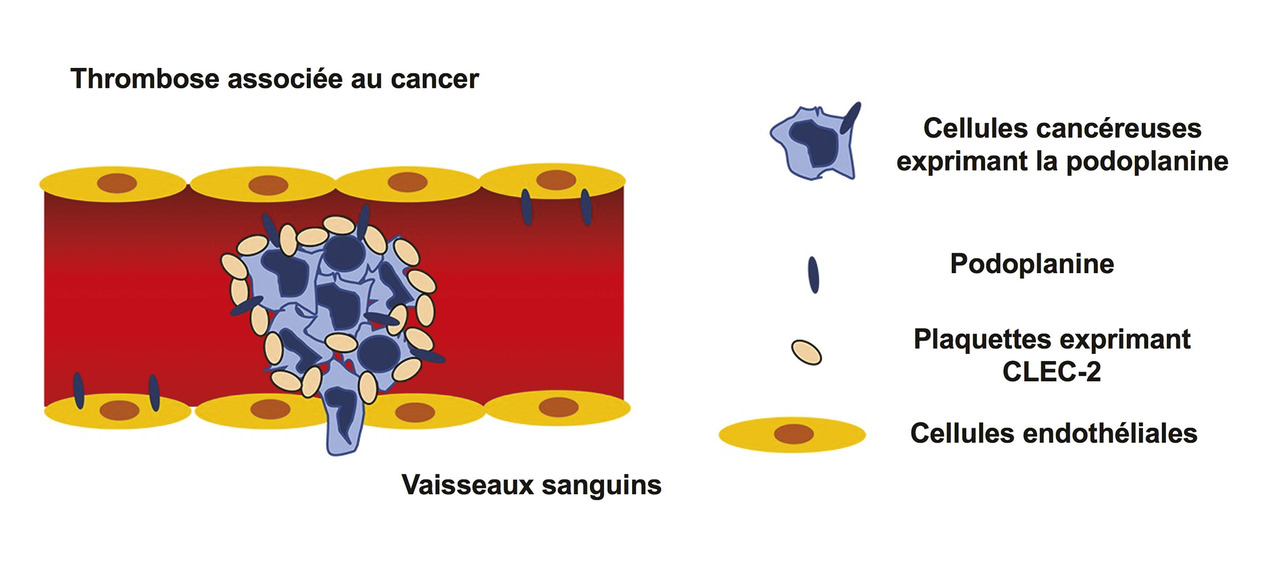
Certaines cellules tumorales peuvent également activer les plaquettes par la podoplanine.3 La podoplanine est une sialoglycoprotéine transmembranaire ligand du récepteur plaquettaire C-type lectin-like receptor 2 (CLEC-2). Elle est présente à la surface de certaines cellules tumorales (mélanome, carcinome épidermoïde, séminome, tumeur cancéreuse cérébrale…) L’augmentation des niveaux de podoplanine est associée à la présence de métastases tumorales ou à la progression maligne ; la podoplanine provenant notamment de tumeurs cérébrales active les plaquettes circulantes, et le risque thrombotique est accru chez ces patients (
Activation leucocytaire
Les polynucléaires neutrophiles (PNN) activés par les cellules tumorales exposent à leur surface des niveaux élevés de FT ainsi que des récepteurs pour les molécules d’adhésion des plaquettes et des cellules endothéliales ; cela a été bien démontré dans le cas des néoplasmes myéloprolifératifs.
Les cellules cancéreuses prédisposent les neutrophiles à la libération de NETs (neutrophils extracellular traps). Les NETs proviennent de l’externalisation de l’ADN (nucléaire ou mitochondrial) libéré du noyau des neutrophiles, associé à des histones et des protéases granulaires, après activation des PNN. Ces NETs sont responsables d’une adhésion et d’une activation plaquettaire ainsi que d’une activation de la phase-contact de la coagulation,4 à l’origine de l’effet prothrombotique (
Activation de l'endothélium
Parmi les cytokines relarguées par les cellules tumorales, l’interleukine-1bêta et le facteur de nécrose tumorale augmentent l’expression du TF et régulent notamment l’expression de la thrombomoduline (TM) à la surface endothéliale.
La TM est un récepteur membranaire des cellules endothéliales vasculaires qui forme un complexe avec la thrombine pour activer la protéine C.
Chez les patients atteints de cancer, on observe des niveaux accrus de TM soluble et une expression réduite de la TM de surface, aboutissant à une perte d’activité anticoagulante de l’endothélium.
Des mécanismes mieux connus et voisins de ceux de la tumorigenèse
La thrombose associée au cancer est un problème clinique majeur car les événements thrombotiques augmentent la morbidité et la mortalité des patients. On connaît aujourd’hui de mieux en mieux les mécanismes d’hypercoagulabilité qui prédisposent aux complications thrombotiques. Ils sont souvent identiques
à ceux de la tumorogenèse, permettant la croissance et la survie des tumeurs.
1. Falanga A, Russo L, Milesi V, Vignoli A. Mechanisms and risk factors of thrombosis in cancer. Crit Rev Oncol Hematol 2017;118:79-83.
2. Del Conde I, Bharwani LD, Dietzen DJ, Pendurthi U, Thiagarajan P, López JA. Microvesicle-associated tissue factor and Trousseau’s syndrome. J Thromb Haemost 2007;5:70-4.
3. Suzuki-Inoue K. Platelets and cancer-associated thrombosis: focusing on the platelet receptor CLEC-2 and podoplanin. Blood 2019;134:1912-8.
4. Demers M, Krause DS, Schatzber D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA 2012;109:13076-81.