Une femme de 65 ans consulte en service d’urgence pour une altération de l’état général évoluant depuis trois semaines, associée à une symptomatologie fonctionnelle digestive de type anorexie, nausées et diarrhées, avec une nette aggravation durant les derniers jours. Elle a comme antécédents une tuberculose pulmonaire et une thyroïdite de Hashimoto traitée par lévothyroxine (100 µg/j).
À l’examen clinique, la pression artérielle est à 100/60 mmHg et la fréquence cardiaque à 80 batt/min. Il existe un pli cutané. L’examen neurologique et la palpation abdominale sont normaux. Un bilan biologique est effectué, objectivant une créatinémie à 12 mg/L (106 μmol/L), une hyponatrémie à 114 mmol/L et une hyperkaliémie à 5,3 mmol/L.
La prise en charge initiale a consisté en une administration de résine échangeuse de potassium et une hydratation par 1 L de sérum salé isotonique sur vingt-quatre heures. Le bilan complémentaire a révélé une natriurèse inadaptée à 77 mmol/24 h en regard d’une natrémie de contrôle à 126 mmol/L, une kaliémie à 4,9 mmol/L. Le cortisol et l’hormone adrénocorticotrope (ACTH) plasmatiques, mesurés au réveil, étaient respectivement à 5,5 µg/dL, soit 152 nmol/L (normale : 9-22 µg/dL) et 432 pg/mL, soit 95 pmol/L (normale < 46 pg/mL).
Un traitement par hémisuccinate d’hydrocortisone 100 mg/24 h en intraveineux après un bolus de 100 mg a alors été initié ; il a permis une régression rapide de la symptomatologie et une correction des troubles hydroélectrolytiques.
La patiente a ensuite été transférée en service d’endocrinologie. Un déficit en minéralocorticoïde associé a été mis en évidence : aldostérone indosable ; activité rénine plasmatique à 8,98 ng/mL/h (normale : 0,3-1,8). Au scanner surrénalien, les surrénales apparaissaient fines et non calcifiées. La positivité des anticorps anti-21-hydroxylase à 1 360 U/mL a confirmé l’origine auto-immune. Un relais par hydrocortisone per os avec une décroissance progressive associé à de la fludrocortisone a été instauré. L’éducation thérapeutique a été débutée pendant l’hospitalisation.
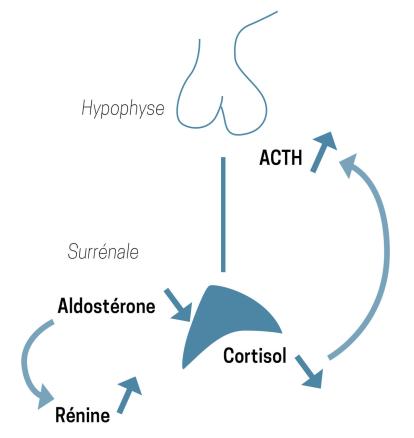
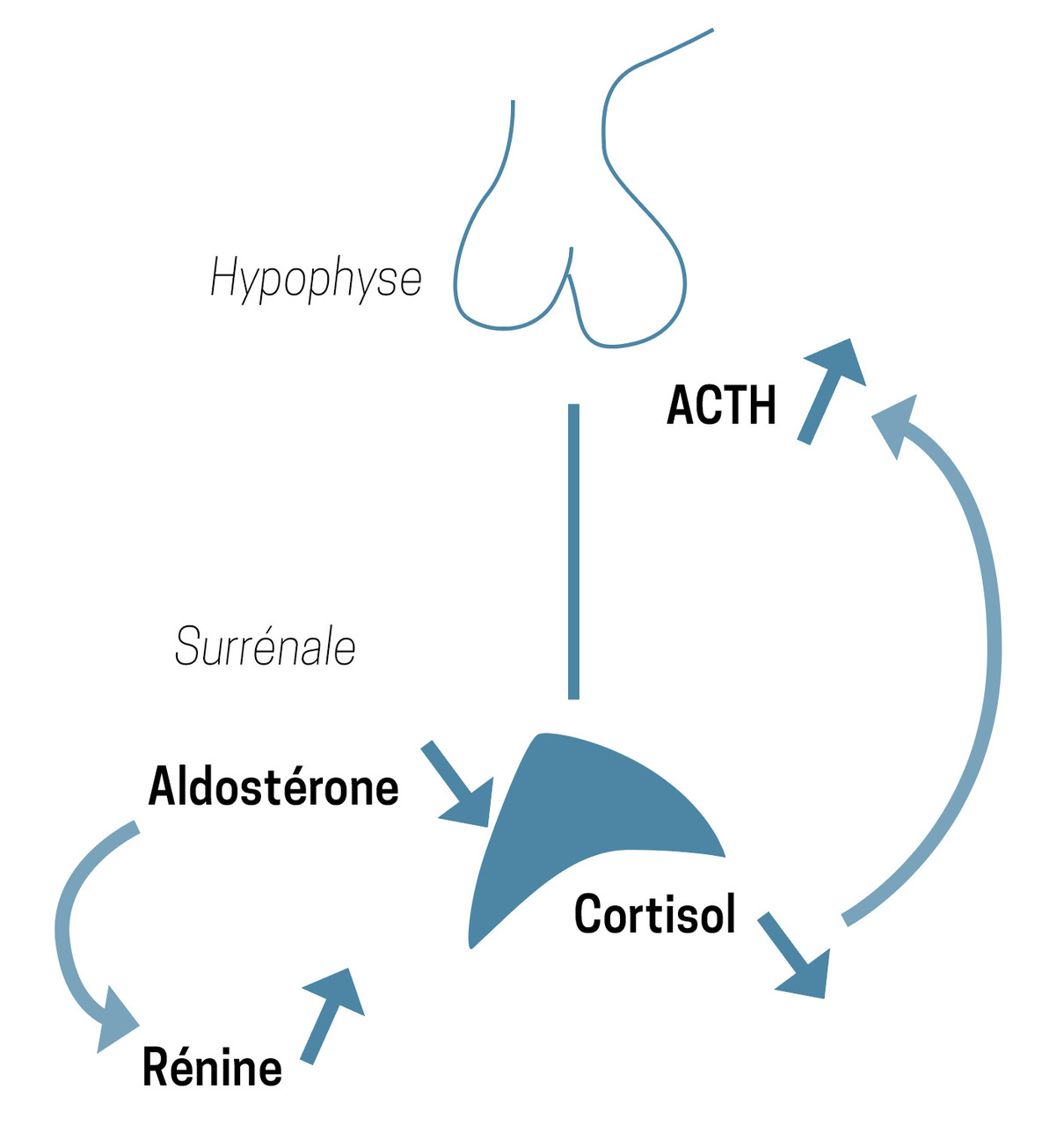
L’insuffisance surrénalienne périphérique (ISP) est une affection rare dont la prévalence est estimée à 10 cas pour 100 000 habitants.1 Elle est liée à une atteinte de la corticosurrénale entraînant un défaut de sécrétion des glucocorticoïdes souvent associé à un défaut de sécrétion minéralocorticoïde (
Sur le plan clinique, l’installation lente et insidieuse ainsi que le caractère peu spécifique des symptômes (
En cas d’ISA en particulier, l’hyponatrémie – liée à une perte rénale en sel2,3 – s’associe à une hyperkaliémie, ce qui doit faire évoquer son diagnostic.2 Toutefois, l’hyperkaliémie peut être limitée en cas de diarrhées profuses ou de vomissements incoercibles.3
Sur le plan étiologique, chez l’adulte, la rétraction corticale auto-immune est désormais la cause la plus fréquente d’ISP, tandis que la tuberculose (dans sa forme extrapulmonaire) ne correspond qu’à moins de 5 % des cas en Europe depuis les années 2000.3 Le caractère auto-immun de la maladie est confirmé par la positivité des anticorps anti-21-hydroxylase.3 Les autres causes sont iatrogènes (surrénalectomie bilatérale, médicament anticortisolique), liées à une nécrose ou une hémorragie bilatérale des surrénales (d’origine traumatique, en lien avec des anticoagulants ou une coagulopathie), infectieuses et, enfin, génétiques, les plus fréquentes dans l’enfance (hyperplasie congénitale des surrénales).2-4
Le traitement de l’ISA est une urgence et doit se faire dès qu’elle est suspectée, si possible après un prélèvement « à la volée » pour un dosage du cortisol, voire de l’ACTH.2 Le cortisol, s’il n’est pas effondré, est bas, ce qui est particulièrement inadapté dans une situation de stress durant laquelle les valeurs devraient être au contraire augmentées.2 L’ACTH est élevée, souvent à plus de 5 fois la normale.2 Contrairement au cas présenté ici, il ne faut pas retarder le début du traitement pour réaliser les dosages hormonaux à 8 h ou attendre les résultats des examens complémentaires pour traiter.1,2 Compte tenu de la demi-vie courte de l’hydrocortisone, il est toujours possible de réaliser le dosage du cortisol ultérieurement pour confirmer le diagnostic ; par ailleurs, l’ACTH reste, dans la majorité des cas, élevée chez le patient en ISP, même sous traitement.2 Le traitement initial consiste en une injection parentérale d’hémisuccinate d’hydrocortisone à 100 mg suivie immédiatement d’une seconde injection de 100 mg en continu sur vingt-quatre heures.1 La réponse clinique et biologique rapide après l’instauration de cette supplémentation constitue un véritable test thérapeutique.1 La correction des troubles hydroélectrolytiques inclut l’apport de chlorure de sodium et de soluté glucosé.1,2 L’hyperkaliémie et l’hyponatrémie sont corrigées par l’administration d’hydrocortisone qui, notamment à forte dose, a un effet minéralocorticoïde.1,3
Le traitement au long cours de l’ISP consiste en la prise orale d’hydrocortisone (15 à 25 mg/j).4 Compte tenu de sa demi-vie courte (environ 4 heures) et pour essayer de se rapprocher de la sécrétion physiologique, le traitement par hydrocortisone est instauré en deux ou trois prises. Le déficit en minéralocorticoïde est supplémenté par de la fludrocortisone en une prise le matin (


1. Cortet C, Barat P, Zenaty D, et al. Group 5: Acute adrenal insufficiency in adults and pediatric patients. Ann Endocrinol 2017;78(6):535‑43.
2. Chanson P, Guignat L, Goichot B, et al. Group 2: Adrenal insufficiency: screening methods and confirmation of diagnosis. Ann Endocrinol 2017;78(6):495‑511.
3. Husebye ES, Pearce SH, Krone NP, et al. Adrenal insufficiency. Lancet 2021;397(10274):613‑29.
4. Castinetti F, Guignat L, Bouvattier C, et al. Group 4: Replacement therapy for adrenal insufficiency. Ann Endocrinol 2017;78(6):525‑34.
Une question, un commentaire ?