Nous avons exposé précédemment les premiers pas ayant conduit au développement de deux méthodes de séquençage des molécules d’ADN par le groupe de Frédérick Sanger au Medical Research Council (Cambridge, Angleterre) et de Wally Gilbert à l’université de Harvard (Cambridge, Massachusetts).* Dans ce second volet est décrit l’explosion de nos capacités d’analyse ayant amené au séquençage du génome humain et de milliers d’autres génomes ainsi que de ceux de nombreux virus à ARN, ayant transformé notre vision de la biologie.
L’accélération quasi inattendue des résultats, tels que rapportés dans la 1re partie de cet article (Rev Prat 2021;71:109-14) est liée au développement en parallèle de nombreuses méthodes qui ont permis grâce à celles de Frederick Sanger1 et de Maxam-Gilbert2 de développer d’extraordinaires capacités analytiques, aboutissant lorsque menées à leur terme à la connaissance, avec une exactitude rarement égalée en biologie, de la structure primaire de l’ADN et ce faisant des ARN et protéines qui en découlent.
D’autres avancées spectaculaires
Outre le développement du système de clonage universel de J. Messing en 1980, également cité, et qui facilita grandement l’utilisation de la méthode des terminaisons de chaînes (ou méthode aux didéoxynucléotides [ddN], v. 1re partie), il convient de citer quelques avancées spectaculaires s’ajoutant à la découverte et à la mise à disposition de tous, d’enzymes de restriction offrant des possibilités d’hydrolyse précise et nombreuse de l’ADN.
C’est dans les années 1980 que Marvin Caruthers, chimiste américain, développa une méthode puissante de synthèse in vitro d’oligodéoxynucléotides de séquence définie.3 Cette méthode consiste à ajouter sur un support solide sur lequel est fixé un premier dérivé nucléotidique un deuxième, puis un troisième et ainsi de suite selon la séquence nucléotidique désirée, avec entre chaque étape une série de réactifs appropriés puis de récupérer in fine un oligodéoxynucléotide qui pourra être utilisé comme amorce pour déterminer avec la méthode des terminaisons de chaînes (ou méthode aux ddN [v. 1re partie]) la séquence du fragment cloné dans un vecteur M13 ou pUC. Un peu plus tard, cette méthode de synthèse fut automatisée et des synthétiseurs d’oligdéoxyonucléotides furent disponibles dans les laboratoires de biologie moléculaire.
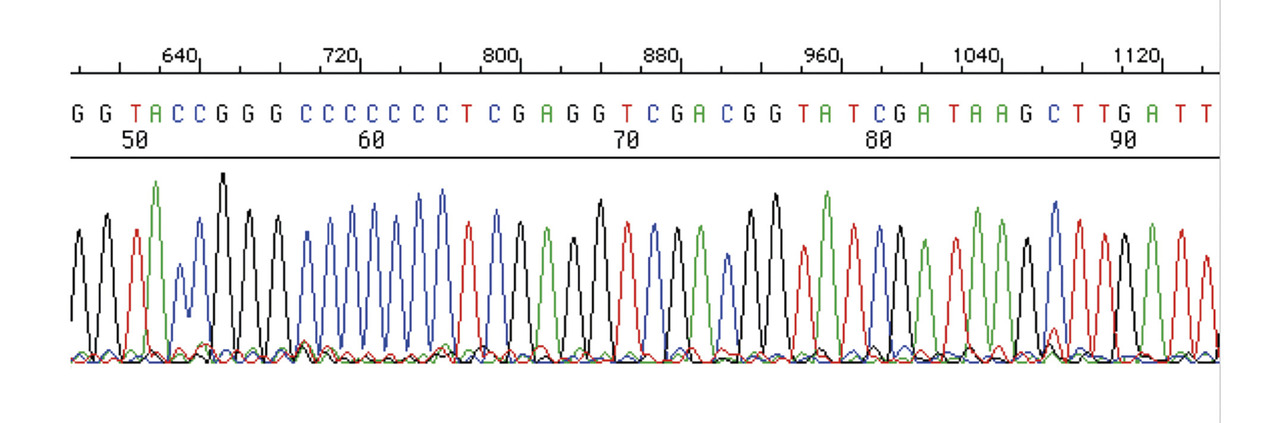
Par ailleurs, Marvin Caruthers créa, avec Leroy Hood, la société Applied Biosystem à l’origine d’un séquenceur automatique d’ADN. Sa particularité était de permettre la migration en parallèle de plusieurs réactions de séquences obtenues par la méthode aux ddN. Pour cela, le marquage au 32P, primitivement utilisé, était remplacé par des fluorophores distincts pour chacune des 4 bases.4 Après analyse, les séquenceurs fournissaient les séquences sous la forme d’un fichier importable dans un ordinateur pour être analysés par les différents programmes développés en parallèle. L’ère du séquençage de l’ADN devenait peu à peu semi-industrielle (fig. 1 à 3 ).
En 1986, Kary Mullis inventa la méthode de polymérisation en chaîne ou PCR qui permit, avec l’utilisation d’amorces spécifiques, d’amplifier et de cloner facilement n’importe quel gène ou fragment de génome.5 Lui aussi, comme beaucoup dans cette aventure, fut honoré d’un prix Nobel en 1993. Michael Smith, chimiste canadien, prix Nobel 1993, mériterait aussi d’être cité pour le développement d’une méthode de mutagenèse dirigée de fragment d’ADN inséré dans un plasmide.
Bien que beaucoup moins célèbre, il apparaît nécessaire de clore cette liste incomplète en nommant Rodger Staden. Ingénieur dans le groupe de Frederick Sanger, Staden développa le premier plusieurs algorithmes de manipulation, de traitement et d’interprétation des séquences nucléotidiques, sans lesquels rien ne pourrait être fait.6 À cet égard il convient de rappeler qu’à l’époque du séquençage du génome du virus de l’hépatite B (VHB), la bio-informatique n’existait pas. Lors de la lecture visuelle des autoradiogrammes, il convenait d’écrire manuellement sur de grands cartons à petits carreaux la séquence des différents fragments de restriction jusqu’à l’obtention d’une séquence continue correspondant à celle des deux chaînes dont la parfaite complémentarité assurait l’exactitude du résultat. Dans le cadre du séquençage du VHB, mes relations personnelles avec le laboratoire de Sanger me permirent d’aller à Cambridge bénéficier de ces travaux. Ainsi la séquence fut entrée manuellement dans l’ordinateur et plusieurs programmes développés par Staden permirent en quelques minutes d’obtenir une multitude d’informations dont la séquence de toutes les protéines codées par ce génome, qu’elles soient hypothétiques ou réelles comme l’antigène HBs. L’ère de la bio-informatique était ouverte et ne se refermerait pas, elle prendrait une importance de plus en plus grande dans l’évolution technologique apportée par les new genome sequencing (NGS) et les interprétations des séquences toujours plus longues et nombreuses.
En parallèle à toutes ces innovations, il convient d’ajouter le développement du fractionnement aléatoire ou « shotgun » de l’ADN à séquencer d’abord par l’usage de la DNase 1 puis ultérieurement par sonication ménagée.7, 8 Cette approche consiste à partir d’un cosmide ou d’un YAC recombinants d’obtenir un grand nombre de fragments de quelques centaines de nucléotides puis de cloner ces derniers dans un des vecteurs de clonage universel développés par Messing.9 Après étalement des clones sur boite de Pétri ces derniers peuvent être séquencés. Ce procédé utilisé universellement permet de s’affranchir de l’usage de beaucoup d’enzymes de restriction dont le hasard de la distribution de leurs sites sensibles ne permet pas toujours l’accès à certaines régions d’un cosmide.
C’est dans les années 1980 que Marvin Caruthers, chimiste américain, développa une méthode puissante de synthèse in vitro d’oligodéoxynucléotides de séquence définie.3 Cette méthode consiste à ajouter sur un support solide sur lequel est fixé un premier dérivé nucléotidique un deuxième, puis un troisième et ainsi de suite selon la séquence nucléotidique désirée, avec entre chaque étape une série de réactifs appropriés puis de récupérer in fine un oligodéoxynucléotide qui pourra être utilisé comme amorce pour déterminer avec la méthode des terminaisons de chaînes (ou méthode aux ddN [v. 1re partie]) la séquence du fragment cloné dans un vecteur M13 ou pUC. Un peu plus tard, cette méthode de synthèse fut automatisée et des synthétiseurs d’oligdéoxyonucléotides furent disponibles dans les laboratoires de biologie moléculaire.
Par ailleurs, Marvin Caruthers créa, avec Leroy Hood, la société Applied Biosystem à l’origine d’un séquenceur automatique d’ADN. Sa particularité était de permettre la migration en parallèle de plusieurs réactions de séquences obtenues par la méthode aux ddN. Pour cela, le marquage au 32P, primitivement utilisé, était remplacé par des fluorophores distincts pour chacune des 4 bases.4 Après analyse, les séquenceurs fournissaient les séquences sous la forme d’un fichier importable dans un ordinateur pour être analysés par les différents programmes développés en parallèle. L’ère du séquençage de l’ADN devenait peu à peu semi-industrielle (
En 1986, Kary Mullis inventa la méthode de polymérisation en chaîne ou PCR qui permit, avec l’utilisation d’amorces spécifiques, d’amplifier et de cloner facilement n’importe quel gène ou fragment de génome.5 Lui aussi, comme beaucoup dans cette aventure, fut honoré d’un prix Nobel en 1993. Michael Smith, chimiste canadien, prix Nobel 1993, mériterait aussi d’être cité pour le développement d’une méthode de mutagenèse dirigée de fragment d’ADN inséré dans un plasmide.
Bien que beaucoup moins célèbre, il apparaît nécessaire de clore cette liste incomplète en nommant Rodger Staden. Ingénieur dans le groupe de Frederick Sanger, Staden développa le premier plusieurs algorithmes de manipulation, de traitement et d’interprétation des séquences nucléotidiques, sans lesquels rien ne pourrait être fait.6 À cet égard il convient de rappeler qu’à l’époque du séquençage du génome du virus de l’hépatite B (VHB), la bio-informatique n’existait pas. Lors de la lecture visuelle des autoradiogrammes, il convenait d’écrire manuellement sur de grands cartons à petits carreaux la séquence des différents fragments de restriction jusqu’à l’obtention d’une séquence continue correspondant à celle des deux chaînes dont la parfaite complémentarité assurait l’exactitude du résultat. Dans le cadre du séquençage du VHB, mes relations personnelles avec le laboratoire de Sanger me permirent d’aller à Cambridge bénéficier de ces travaux. Ainsi la séquence fut entrée manuellement dans l’ordinateur et plusieurs programmes développés par Staden permirent en quelques minutes d’obtenir une multitude d’informations dont la séquence de toutes les protéines codées par ce génome, qu’elles soient hypothétiques ou réelles comme l’antigène HBs. L’ère de la bio-informatique était ouverte et ne se refermerait pas, elle prendrait une importance de plus en plus grande dans l’évolution technologique apportée par les new genome sequencing (NGS) et les interprétations des séquences toujours plus longues et nombreuses.
En parallèle à toutes ces innovations, il convient d’ajouter le développement du fractionnement aléatoire ou « shotgun » de l’ADN à séquencer d’abord par l’usage de la DNase 1 puis ultérieurement par sonication ménagée.7, 8 Cette approche consiste à partir d’un cosmide ou d’un YAC recombinants d’obtenir un grand nombre de fragments de quelques centaines de nucléotides puis de cloner ces derniers dans un des vecteurs de clonage universel développés par Messing.9 Après étalement des clones sur boite de Pétri ces derniers peuvent être séquencés. Ce procédé utilisé universellement permet de s’affranchir de l’usage de beaucoup d’enzymes de restriction dont le hasard de la distribution de leurs sites sensibles ne permet pas toujours l’accès à certaines régions d’un cosmide.
Le programme « génome humain » (1989)
Toutes ces découvertes furent les avancées nécessaires à l’élaboration du Human Genome Project, qui au moment de sa création tenait plus du rêve que de la réalité, tant les méthodes à la disposition de la communauté scientifique étaient encore balbutiantes au regard à l’énormité de la tâche.
La première initiative dans cette direction fut celle de Jean Dausset, prix Nobel de médecine 1980 pour ses travaux sur le complexe majeur d’histocompatibilité (CMH) qui, avec Daniel Cohen, médecin biologiste, créa en 1984 le Centre d’étude du polymorphisme humain (CEPH) pour étudier ce qu’ils avaient pressenti très vite, à savoir l’importance du polymorphisme génétique dans les maladies complexes en particulier. Mais l’idée d’un vaste programme de séquençage du génome humain fut avancée en 1985 par Robert Sinsheimer qui organisa à Santa Cruz (Californie) une réunion à laquelle participèrent plusieurs scientifiques éminents pour discuter de sa faisabilité. Toutefois le projet, par manque de financement, fut mis sous le boisseau. L’année suivante, en 1986, la publication par Renalto Dulbecco dans Science de l’article A turning point in cancer research : sequencing the human genome dont il a été fait mention plus haut à propos du séquençage de la levure (v. 1e partie)10 convainquit le DOE (department of Energy) et son directeur de la biologie Charles de Risi débloqua un budget pour une étude de faisabilité.
En 1988, c’est à l’initiative de Sydney Brenner, qui en lança l’idée au cours du premier congrès consacré à la cartographie et au séquençage des génomes qui eut lieu au Cold Spring Harbor Laboratory (CSHL), que fut créé la Human Genome Organization (HUGO) réunissant à ces débuts quelque 17 pays et 42 scientifiques, chiffres qui augmentèrent beaucoup au cours des années. Cette initiative fut suivie par la création en 1989 du programme « The Human Genome Project » dont James Watson fut le premier directeur et l’octroi par le National Institute of Health (NIH) d’un budget initial de 3 milliards de dollars pour le mettre en œuvre.
La première initiative dans cette direction fut celle de Jean Dausset, prix Nobel de médecine 1980 pour ses travaux sur le complexe majeur d’histocompatibilité (CMH) qui, avec Daniel Cohen, médecin biologiste, créa en 1984 le Centre d’étude du polymorphisme humain (CEPH) pour étudier ce qu’ils avaient pressenti très vite, à savoir l’importance du polymorphisme génétique dans les maladies complexes en particulier. Mais l’idée d’un vaste programme de séquençage du génome humain fut avancée en 1985 par Robert Sinsheimer qui organisa à Santa Cruz (Californie) une réunion à laquelle participèrent plusieurs scientifiques éminents pour discuter de sa faisabilité. Toutefois le projet, par manque de financement, fut mis sous le boisseau. L’année suivante, en 1986, la publication par Renalto Dulbecco dans Science de l’article A turning point in cancer research : sequencing the human genome dont il a été fait mention plus haut à propos du séquençage de la levure (v. 1e partie)10 convainquit le DOE (department of Energy) et son directeur de la biologie Charles de Risi débloqua un budget pour une étude de faisabilité.
En 1988, c’est à l’initiative de Sydney Brenner, qui en lança l’idée au cours du premier congrès consacré à la cartographie et au séquençage des génomes qui eut lieu au Cold Spring Harbor Laboratory (CSHL), que fut créé la Human Genome Organization (HUGO) réunissant à ces débuts quelque 17 pays et 42 scientifiques, chiffres qui augmentèrent beaucoup au cours des années. Cette initiative fut suivie par la création en 1989 du programme « The Human Genome Project » dont James Watson fut le premier directeur et l’octroi par le National Institute of Health (NIH) d’un budget initial de 3 milliards de dollars pour le mettre en œuvre.
Le nématode C. elegans (1998)
C’est dans ce contexte qu’était publiée en 1998 la séquence du génome d’un ver, le nématode Caenorhabditis elegans,11 premier organisme multicellulaire que Brenner avait choisi depuis plusieurs années comme modèle pour la biologie du développement.
La partie codante de ce génome (97 mégabases répartis dans 5 autosomes et le chromosome X) révélait quelque 19 099 phases de lecture (ORF) ouvertes dont la traduction en séquences protéiques montait que 20 % d’entre elles et 40 % des ORF de levure seraient des orthologues, c’est-à-dire des gènes qui auraient été hérité d’un ancêtre commun renforçant par la même l’intérêt s’il en était besoin du séquençage des génomes source d’information importante dans le transfert des connaissances d’un organisme vers un autre.
Le séquençage fut réalisé à partir de banques de cosmides acceptant des inserts de 50 Kb environ et à la fin de YAC acceptant de plus grands inserts mieux adaptés à la taille du génome. Ces clones furent ordonnés avec beaucoup de soin afin de couvrir la quasi-totalité du génome, y compris des télomères dans lesquels la présence de grande répétition rend l’analyse particulièrement délicate et des centromères dont la structure répétitive est beaucoup moins marquée chez Caenorhabditis elegans. Les clones sélectionnés à partir des cartes physiques furent analysés après fractionnement mécanique aléatoire, clonage dans le phage M13 ou un plasmide et séquencés avec la méthode des terminaisons de chaîne.1 Les réactions de séquence furent réalisées avec des amorces fluorescentes ou, mieux, des ddN fluorescents. Après migration et lecture automatique des réactions de séquences avec des séquenceurs Applied Biosystems utilisant un algorithme dédié appelé PHRED,12 les séquences plus ou moins longues d’environ 150 à 200 nt à l’époque étaient ensuite assemblées en contigs (séquence génomique continue) avec PHRAP.13 Le comblement des trous et la résolution des ambiguïtés étaient obtenus avec deux autres algorithmes (GAP et CONSED).14 Fait remarquable pour un génome de cette taille, l’assemblage final fournit une séquence en seulement 8 contigs pour 6 autosomes et le chromosome X, c’est-à-dire que la majorité des chromosomes étaient représentés par une séquence continue d’un télomère à l’autre, démontrant par là-même l’importance du choix des clones recombinants à séquencer et donc d’une carte préalable robuste.
La communauté scientifique en raison de son organisation éclatée en de très nombreux laboratoires, et vraisemblablement encouragée par les bons résultats obtenus avec la levure de boulanger et le ver nématode, privilégia une approche similaire clone par clone. Les différents laboratoires du consortium se partagèrent les chromosomes et construisirent des cartes physiques de cosmides et de YAC recombinants. La séquence nucléotidique des clones sélectionnés à partir des cartes fut déterminée de façon similaire à ceux de la levure et du ver. Ici encore, l’utilisation des différents algorithmes de traitement des données fut indispensable à la détermination de la séquence publiée.15
C’est cette stratégie efficace mais relativement lente qui fut suivie par le consortium de laboratoires jusqu’à l’intrusion dans le domaine de Craig Venter. Ce dernier chercheur au NIH, avait activement participé au développement des EST (expressed sequence tags) qui correspondent à des fragments clonés plus ou moins longs d’ARN messagers produits par un tissu donné et qui permettent de faire l’inventaire des gènes exprimés. En 1992, Venter quitta le NIH à la suite du refus de ce dernier de breveter toutes ces séquences et monta une fondation, The Institute for Genome Research (TIGR), dotée d’une plateforme de séquençage à grande échelle avec l’aide d’H. Smith, déjà cité à propos des ER.
En 1995, le groupe publia la séquence complète du génome d’Haemophilus influenzae,16 premier organisme dont la séquence du génome fut ainsi déterminée (1 830 140 pb codant 1 740 gènes). À la suite de cet exploit, il fonda la société Celera, avec l’aide de la société Perkin-Elmer, avec l’objectif de séquencer indépendamment du consortium international l’intégralité du génome humain.
La partie codante de ce génome (97 mégabases répartis dans 5 autosomes et le chromosome X) révélait quelque 19 099 phases de lecture (ORF) ouvertes dont la traduction en séquences protéiques montait que 20 % d’entre elles et 40 % des ORF de levure seraient des orthologues, c’est-à-dire des gènes qui auraient été hérité d’un ancêtre commun renforçant par la même l’intérêt s’il en était besoin du séquençage des génomes source d’information importante dans le transfert des connaissances d’un organisme vers un autre.
Le séquençage fut réalisé à partir de banques de cosmides acceptant des inserts de 50 Kb environ et à la fin de YAC acceptant de plus grands inserts mieux adaptés à la taille du génome. Ces clones furent ordonnés avec beaucoup de soin afin de couvrir la quasi-totalité du génome, y compris des télomères dans lesquels la présence de grande répétition rend l’analyse particulièrement délicate et des centromères dont la structure répétitive est beaucoup moins marquée chez Caenorhabditis elegans. Les clones sélectionnés à partir des cartes physiques furent analysés après fractionnement mécanique aléatoire, clonage dans le phage M13 ou un plasmide et séquencés avec la méthode des terminaisons de chaîne.1 Les réactions de séquence furent réalisées avec des amorces fluorescentes ou, mieux, des ddN fluorescents. Après migration et lecture automatique des réactions de séquences avec des séquenceurs Applied Biosystems utilisant un algorithme dédié appelé PHRED,12 les séquences plus ou moins longues d’environ 150 à 200 nt à l’époque étaient ensuite assemblées en contigs (séquence génomique continue) avec PHRAP.13 Le comblement des trous et la résolution des ambiguïtés étaient obtenus avec deux autres algorithmes (GAP et CONSED).14 Fait remarquable pour un génome de cette taille, l’assemblage final fournit une séquence en seulement 8 contigs pour 6 autosomes et le chromosome X, c’est-à-dire que la majorité des chromosomes étaient représentés par une séquence continue d’un télomère à l’autre, démontrant par là-même l’importance du choix des clones recombinants à séquencer et donc d’une carte préalable robuste.
La communauté scientifique en raison de son organisation éclatée en de très nombreux laboratoires, et vraisemblablement encouragée par les bons résultats obtenus avec la levure de boulanger et le ver nématode, privilégia une approche similaire clone par clone. Les différents laboratoires du consortium se partagèrent les chromosomes et construisirent des cartes physiques de cosmides et de YAC recombinants. La séquence nucléotidique des clones sélectionnés à partir des cartes fut déterminée de façon similaire à ceux de la levure et du ver. Ici encore, l’utilisation des différents algorithmes de traitement des données fut indispensable à la détermination de la séquence publiée.15
C’est cette stratégie efficace mais relativement lente qui fut suivie par le consortium de laboratoires jusqu’à l’intrusion dans le domaine de Craig Venter. Ce dernier chercheur au NIH, avait activement participé au développement des EST (expressed sequence tags) qui correspondent à des fragments clonés plus ou moins longs d’ARN messagers produits par un tissu donné et qui permettent de faire l’inventaire des gènes exprimés. En 1992, Venter quitta le NIH à la suite du refus de ce dernier de breveter toutes ces séquences et monta une fondation, The Institute for Genome Research (TIGR), dotée d’une plateforme de séquençage à grande échelle avec l’aide d’H. Smith, déjà cité à propos des ER.
En 1995, le groupe publia la séquence complète du génome d’Haemophilus influenzae,16 premier organisme dont la séquence du génome fut ainsi déterminée (1 830 140 pb codant 1 740 gènes). À la suite de cet exploit, il fonda la société Celera, avec l’aide de la société Perkin-Elmer, avec l’objectif de séquencer indépendamment du consortium international l’intégralité du génome humain.
La mouche du vinaigre (2000)
Conforté par ses premiers résultats avec le séquençage du génome d’Haemophilus influenzae et à titre de rodage, C. Venter choisit le génome de la drosophile dont il détermina la quasi-intégralité de la partie euchromatique (120 mégabases sur les 180 Gb estimée de la totalité du génome). Bien qu’avec des imperfections et des lacunes, à la différence de toutes les autres séquences publiées jusque-là, mais en un temps record, cette séquence révélait la présence d’environ 13 600 gènes, soit un peu moins que dans le génome de C. elegans pourtant de taille plus petite.17
Au lieu de construire des banques de cosmides ou de YAC puis de les cartographier, l’ADN de drosophile fut fractionné en une seule étape en milliers de fragments puis clonés. Les dizaines de milliers de lectures de séquences de ces clones furent alors assemblées par l’utilisation de puissants algorithmes.
Fort de l’expérience acquise Celera attaqua le séquençage du génome humain de la même façon, c’est-à-dire qu’en une seule étape la totalité d’un échantillon d’ADN humain fut fractionné, cloné et les milliers de clones séquencés en parallèle dans des séquenceurs Applied Biosystems mettant à profit de façon industrielle la méthode aux ddN.1
Au lieu de construire des banques de cosmides ou de YAC puis de les cartographier, l’ADN de drosophile fut fractionné en une seule étape en milliers de fragments puis clonés. Les dizaines de milliers de lectures de séquences de ces clones furent alors assemblées par l’utilisation de puissants algorithmes.
Fort de l’expérience acquise Celera attaqua le séquençage du génome humain de la même façon, c’est-à-dire qu’en une seule étape la totalité d’un échantillon d’ADN humain fut fractionné, cloné et les milliers de clones séquencés en parallèle dans des séquenceurs Applied Biosystems mettant à profit de façon industrielle la méthode aux ddN.1
Le brouillon du génome humain (2001)
La publication la même semaine dans Nature15 et Science18 fut l’objet de discussions âpres, tant l’animosité entre les deux groupes avec la suspicion de piratage par le groupe de Celera de données confidentielles du consortium était grande. La publication donna lieu à une cérémonie quelque peu grandiloquente, non dénuée d’arrière-pensée politique, présidée par Bill Clinton, président des États-Unis, et Tony Blair, Premier ministre britannique, eu égard à ce que leurs deux pays avaient séquencé plus de 80 % du génome.
Les deux séquences produites n’étaient certes pas parfaites, il s’agissait tout de même de brouillons de qualité qui permettaient déjà d’avoir de nombreuses et importantes informations telles que le nombre de gènes codant des protéines. Celui-ci était estimé à 25 à 30 000, soit à peine plus que le nombre identifié chez le nématode et deux fois seulement celui de la mouche, alors qu’on s’attendait à un chiffre de l’ordre de 100 000 gènes, ce qui a surpris. Par ailleurs l’ensemble de ces gènes représentait moins de 5 % de la séquence. En revanche ces gènes, très morcelés en exons de petite taille (codant 50 à 100 acides aminés) du fait de la présence de nombreux et larges introns, offrent la possibilité d’agencement (splicing) multiples permettant à un gène de coder plusieurs protéines. Bien d’autres découvertes furent faites, par exemple celles que des centaines de gènes semblaient résulter d’un transfert horizontal à partir des bactéries à un moment ou un autre du phyllum des vertébrés ou qu’environ la moitié du génome était représentée par des éléments transposables devenus inactifs, etc.
Fort de ce résultat plus qu’impressionnant, la communauté poursuivit ses efforts afin d’améliorer ce brouillon et, depuis, plusieurs séquences ont été successivement déposées dans les bases de données comme GENBANK abritée par le National Center for Biotechnology Information (NCBI) ou ENSEMBL qui dépend de European Bioinformatics Institute (EBI) et du Wellcome Trust Sanger Institute, avec un dernier dépôt le 28 février 2019.
Comme on pouvait s’y attendre, tout au long de ces années, des centaines de génomes représentatifs de tous les embranchements du monde vivant ont été séquencés. Toutefois il convient de rappeler que si les séquences obtenues du génome de l’homme, de la souris, du chien et de quelques autres sont de grande qualité, la très grande majorité des séquences de génomes publiées à ce jour s’apparentent plus à des brouillons, dont l’intérêt reste néanmoins très grand, ne serait-ce que par l’inventaire des gènes et leur traduction in silico en protéines et une vision renouvelée de l’évolution. À ce bref panorama, il faut aussi ajouter que la séquence des génomes de plusieurs milliers d’entre nous plus ou moins représentatifs de la diversité des populations humaines a été déterminée ainsi que de celles de nos ancêtres néandertalien19 ou de l’homme de Denisova,20 montrant la présence de plusieurs de leurs gènes dans les populations humaines actuelles, à des degrés différents selon celles-ci, témoignant d’un certain niveau de cohabitation et de croisement inter-espèces.
Les deux séquences produites n’étaient certes pas parfaites, il s’agissait tout de même de brouillons de qualité qui permettaient déjà d’avoir de nombreuses et importantes informations telles que le nombre de gènes codant des protéines. Celui-ci était estimé à 25 à 30 000, soit à peine plus que le nombre identifié chez le nématode et deux fois seulement celui de la mouche, alors qu’on s’attendait à un chiffre de l’ordre de 100 000 gènes, ce qui a surpris. Par ailleurs l’ensemble de ces gènes représentait moins de 5 % de la séquence. En revanche ces gènes, très morcelés en exons de petite taille (codant 50 à 100 acides aminés) du fait de la présence de nombreux et larges introns, offrent la possibilité d’agencement (splicing) multiples permettant à un gène de coder plusieurs protéines. Bien d’autres découvertes furent faites, par exemple celles que des centaines de gènes semblaient résulter d’un transfert horizontal à partir des bactéries à un moment ou un autre du phyllum des vertébrés ou qu’environ la moitié du génome était représentée par des éléments transposables devenus inactifs, etc.
Fort de ce résultat plus qu’impressionnant, la communauté poursuivit ses efforts afin d’améliorer ce brouillon et, depuis, plusieurs séquences ont été successivement déposées dans les bases de données comme GENBANK abritée par le National Center for Biotechnology Information (NCBI) ou ENSEMBL qui dépend de European Bioinformatics Institute (EBI) et du Wellcome Trust Sanger Institute, avec un dernier dépôt le 28 février 2019.
Comme on pouvait s’y attendre, tout au long de ces années, des centaines de génomes représentatifs de tous les embranchements du monde vivant ont été séquencés. Toutefois il convient de rappeler que si les séquences obtenues du génome de l’homme, de la souris, du chien et de quelques autres sont de grande qualité, la très grande majorité des séquences de génomes publiées à ce jour s’apparentent plus à des brouillons, dont l’intérêt reste néanmoins très grand, ne serait-ce que par l’inventaire des gènes et leur traduction in silico en protéines et une vision renouvelée de l’évolution. À ce bref panorama, il faut aussi ajouter que la séquence des génomes de plusieurs milliers d’entre nous plus ou moins représentatifs de la diversité des populations humaines a été déterminée ainsi que de celles de nos ancêtres néandertalien19 ou de l’homme de Denisova,20 montrant la présence de plusieurs de leurs gènes dans les populations humaines actuelles, à des degrés différents selon celles-ci, témoignant d’un certain niveau de cohabitation et de croisement inter-espèces.
Nouvelles méthodes de séquençage ou méthodes de 2e génération
La puissance de la méthode aux ddN de Frederick Sanger grandement améliorée par de nombreux apports successifs a été de plus en plus pratiquée dans des centres dédiés comme le Sanger Institute à Hinxton en Angletererre, le BROAD Institute à Boston ou le Baylor’s Genome center à Houston pour ne citer que ces trois-là, a depuis quelques années en partie été remplacée par ce qu’on a appelé les Next Generation Sequencing (NGS) pour qui, comme nous le verrons, du moins pour les premières, repose sur le même principe que la méthode aux ddN, à savoir l’hybridation d’une amorce oligonucléotidique sur une matrice à séquencer clonée dans un vecteur. En revanche, elle s’en distingue par la grande parallélisation des analyses.
Le développement de ces méthodes débuta dans les années 1990 à l’initiative de plusieurs groupes. Mais ce n’est qu’en 2005 que le premier instrument commercialisé par Roche dénommé Roche 454 fut accessible aux laboratoires. Le principe de la méthode repose sur la fabrication de banques de fragments d’ADN auxquels sont liés à chacune de leurs deux extrémités 3’ deux oligonucléotides A et B sur lesquels seront hybridés des oligonucléotides complémentaires permettant la synthèse d’un brin d’ADN comme dans la méthode de Sanger. Toutefois, avant de procéder à l’amplification, les molécules d’ADN avec tous les réactifs nécessaires sont dispersées dans une solution d’huile de façon à obtenir une émulsion dans laquelle chaque micelle emprisonne une seule molécule créant ainsi des milliers de mini-réacteurs qui sont distribués individuellement dans des microplaques. Lors de l’opération de séquençage proprement dite, un nucléotide est ajouté à la fois et son accrochage éventuel sur la matrice en élongation est détecté par la libération du pyrophosphate et l’émission de fluorescence sous l’action de la luciférase, comme développée primitivement par Ronaghi et Nyrén. 21
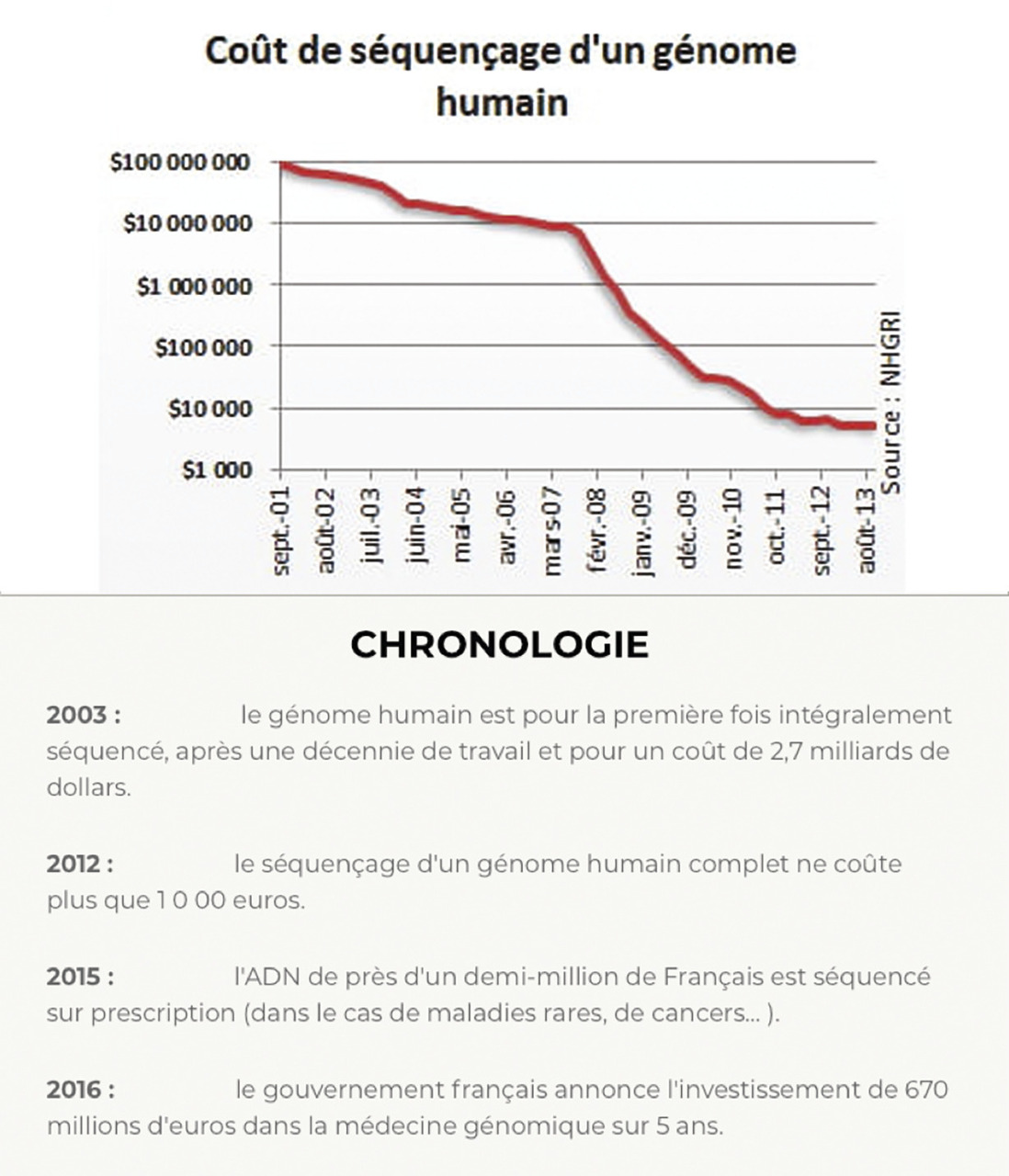
L’addition successive des quatre nucléotides A, G, C, T constituant un cycle qui est répété environ 200 fois permet de définir la séquence à analyser. La puissance de la méthode est de l’ordre de 100 000 séquences déterminées en parallèle. Bien qu’extrêmement puissante, elle n’en fut pas moins assez rapidement dépassée par plusieurs instruments dont en réalité un seul s’est finalement imposé, celui de Solexa racheté par Illumina, qui domina totalement le domaine. Par bien des côtés, la méthode développée par Solexa/Illumina ressemble à la méthode précédente en ce sens qu’après fabrication d’une librairie, celle-ci est déposée dans ce qu’Illumina appelle une « flow cell » dans laquelle chaque molécule d’ADN s’hybride par son extrémité 3’ (à laquelle lors de la constitution de la banque avait été ligaturé un oligonucléotide A) à un oligonucléotide complémentaire B greffé sur la « flow cell » par son extrémité 5’. Comme son nom le laisse penser, différents réactifs sont introduits par la machine dans la flow cell dans laquelle une amplification des molécules se fait, permettant d’obtenir des bouquets de chaque chaîne suivie de la détermination de leurs séquences à partir d’une amorce par ajout séquentiel comme dans la méthode précédente des quatre bases A,G,C,T, chacune porteuse d’un fluorophore différent permettant leur identification. La puissance des derniers séquenceurs Illumina est telle qu’en deux jours la machine est en mesure de déterminer jusqu’à 330 Gb, soit l’équivalent de 100 génomes humains, d’un échantillon unique ou de plusieurs déposés en même temps dans la flow cell et dont le nombre n’est limité que par la profondeur du séquençage que l’on souhaite pour chacun d’eux.22 La puissance d’analyse de ces machines entraîna une baisse très importante des coûts (fig. 3 ).
Le développement de ces méthodes débuta dans les années 1990 à l’initiative de plusieurs groupes. Mais ce n’est qu’en 2005 que le premier instrument commercialisé par Roche dénommé Roche 454 fut accessible aux laboratoires. Le principe de la méthode repose sur la fabrication de banques de fragments d’ADN auxquels sont liés à chacune de leurs deux extrémités 3’ deux oligonucléotides A et B sur lesquels seront hybridés des oligonucléotides complémentaires permettant la synthèse d’un brin d’ADN comme dans la méthode de Sanger. Toutefois, avant de procéder à l’amplification, les molécules d’ADN avec tous les réactifs nécessaires sont dispersées dans une solution d’huile de façon à obtenir une émulsion dans laquelle chaque micelle emprisonne une seule molécule créant ainsi des milliers de mini-réacteurs qui sont distribués individuellement dans des microplaques. Lors de l’opération de séquençage proprement dite, un nucléotide est ajouté à la fois et son accrochage éventuel sur la matrice en élongation est détecté par la libération du pyrophosphate et l’émission de fluorescence sous l’action de la luciférase, comme développée primitivement par Ronaghi et Nyrén. 21
L’addition successive des quatre nucléotides A, G, C, T constituant un cycle qui est répété environ 200 fois permet de définir la séquence à analyser. La puissance de la méthode est de l’ordre de 100 000 séquences déterminées en parallèle. Bien qu’extrêmement puissante, elle n’en fut pas moins assez rapidement dépassée par plusieurs instruments dont en réalité un seul s’est finalement imposé, celui de Solexa racheté par Illumina, qui domina totalement le domaine. Par bien des côtés, la méthode développée par Solexa/Illumina ressemble à la méthode précédente en ce sens qu’après fabrication d’une librairie, celle-ci est déposée dans ce qu’Illumina appelle une « flow cell » dans laquelle chaque molécule d’ADN s’hybride par son extrémité 3’ (à laquelle lors de la constitution de la banque avait été ligaturé un oligonucléotide A) à un oligonucléotide complémentaire B greffé sur la « flow cell » par son extrémité 5’. Comme son nom le laisse penser, différents réactifs sont introduits par la machine dans la flow cell dans laquelle une amplification des molécules se fait, permettant d’obtenir des bouquets de chaque chaîne suivie de la détermination de leurs séquences à partir d’une amorce par ajout séquentiel comme dans la méthode précédente des quatre bases A,G,C,T, chacune porteuse d’un fluorophore différent permettant leur identification. La puissance des derniers séquenceurs Illumina est telle qu’en deux jours la machine est en mesure de déterminer jusqu’à 330 Gb, soit l’équivalent de 100 génomes humains, d’un échantillon unique ou de plusieurs déposés en même temps dans la flow cell et dont le nombre n’est limité que par la profondeur du séquençage que l’on souhaite pour chacun d’eux.22 La puissance d’analyse de ces machines entraîna une baisse très importante des coûts (
Méthodes de troisième génération
En parallèle à tous ces développements, dès les années 1980, plusieurs groupes de chercheurs académiques mais également du privé ont cherché à développer des méthodes de séquençage par lecture directe de l’enchaînement des nucléotides. Dans l’approche développée par Pac. Bio à partir des travaux de Eid et al,23 chaque accrochage d’un nucléotide est détecté et caractérisé par une émission de fluorescence de longueur d’ondes différente pour chaque nucléotide. Dans l’approche développée par Oxford Nanopore Technologies (ONT), ce même principe de lecture directe a été mis en œuvre mais, dans cette approche, le principe développé par Sanger de néo-synthèse d’ADN à partir d’une amorce hybridée sur la chaîne à séquencer est abandonné. Dans ce dernier cas, une des deux chaînes d’ADN de la molécule à analyser est progressivement détachée et forcée à passer dans un pore fait de protéines comme un fil qu’on enfilerait dans un chas d’aiguille créant au passage de chaque nucléotide un courant électrique caractéristique de chacune des quatre bases. Les résultats obtenus par cette approche sont impressionnants par la longueur des lectures qui peuvent atteindre 900 Kb avec la technologie ONT.24
Certes, les taux d’erreurs y sont actuellement supérieurs à ceux obtenus avec la méthode Illumina, mais ces nouvelles approches associées aux précédentes permettent des assemblages de lecture grandement facilités et robustes par la longueur des séquences lues, permettent également d’accéder et de franchir les régions répétitives des génomes de mammifères en particulier. De sorte qu’actuellement la stratégie de séquençage de nouveaux génomes se fait plus facilement en débutant l’analyse par de très longue lecture sur ONT ou PacBio sur lesquelles viennent se positionner les séquences obtenues sur appareil Illumina entraînant un gain de temps très important.
D’autres approches sont également à l’étude mais aucune ne peut prétendre rivaliser avec celles actuellement disponibles
Certes, les taux d’erreurs y sont actuellement supérieurs à ceux obtenus avec la méthode Illumina, mais ces nouvelles approches associées aux précédentes permettent des assemblages de lecture grandement facilités et robustes par la longueur des séquences lues, permettent également d’accéder et de franchir les régions répétitives des génomes de mammifères en particulier. De sorte qu’actuellement la stratégie de séquençage de nouveaux génomes se fait plus facilement en débutant l’analyse par de très longue lecture sur ONT ou PacBio sur lesquelles viennent se positionner les séquences obtenues sur appareil Illumina entraînant un gain de temps très important.
D’autres approches sont également à l’étude mais aucune ne peut prétendre rivaliser avec celles actuellement disponibles
Des développements majeurs dans de multiples domaines
Au cours de ces 50 dernières années, plusieurs méthodes d’analyse de la séquence de l’ADN ont été successivement développées. Celles-ci bénéficiant de nombreuses autres découvertes et du développement sans pareil de l’informatique (qui n’existait pas au début) nous ont offert la capacité insoupçonnable au début d’analyser de nombreux génomes dont le génome humain et d’appréhender la signification biologique de ces enchaînements de nucléotides. Cette capacité d’analyse a profondément bouleversé la biologie et la médecine humaine et vétérinaire non seulement par l’analyse de l’ADN génomique mais aussi par l’analyse des ARN messagers (transcriptome) exprimés par les différents tissus ou organes ainsi que par l’analyse plus récente des divers microbiomes c’est-à-dire des gènes des milliards de bactéries, champignons et autres éléments de la flore que nous abritons, dont peut-être le plus important est le microbiome intestinal sans lequel nous ne pourrions survivre. Le séquençage de l’ADN a bouleversé également beaucoup d’autres domaines comme l’agriculture, l’élevage ou encore l’histoire, la sociologie, la justice et d’autres encore, et peu de domaines lui échappent aujourd’hui. Le mot ADN est même entré dans le langage courant pour évoquer ce qui fait l’essentiel d’une personne, ce qui peut la caractériser dans ses goûts, ses attitudes ou encore son moi, mais aussi, de façon plus surprenante, pour définir le cœur d’une entreprise en évoquant l’ADN de celle-ci. Dans le même temps, personne ne s’étonne des progrès réalisés. Qui aurait pu imaginer il y a peu qu’à peine le SARS-CoV-2 apparu, l’intégralité de son génome ARN, soit 32 682 nt, aurait été déchiffré aussi rapidement et que, depuis, des milliers d’isolats ont été également séquencés permettant de suivre quasiment en temps réel les mutations affectant ce virus à ARN et à anticiper ou identifier l’émergence de variants plus infectieux ou pathogène ?
Les laboratoires d’une petite poignée de pays dont principalement et de loin les États-Unis et le Royaume-Uni ont participé à cette aventure. En France, il faut bien le reconnaître, notre contribution a été modeste au regard de la taille du pays et de son importance dans le domaine scientifique. Malgré des débuts prometteurs, séquençage du génome du VHB, création du CEPH, création du Centre de recherche et d’étude des génomes (GREG), création par le CNRS du Génoscope qui séquença entre autres le chromosome 14 humain, la communauté scientifique du pays ne s’est pas, du moins au début, impliquée dans l’aventure. La raison de cette attitude doit être recherchée dans notre propension à privilégier la partie intellectuelle de la recherche trop souvent au détriment de la technique. Il est peu vraisemblable qu’un chercheur comme Frederick Sanger qui, pendant des années, a essayé de développer une méthode à partir d’un phage monocaténaire ayant au départ peu de chance d’être opérationnelle avec de l’ADN double brin aurait été soutenu financièrement, intellectuellement par son institution. Fort heureusement, devant l’importance prise ces dernières années par l’analyse de l’ADN, les esprits ont quelque peu évolué favorablement.
Il est toujours difficile de prédire l’avenir, toutefois on peut penser que les développements technologiques vont porter plus sur la recherche d’une baisse des coûts, d’une augmentation de la fiabilité des résultats qui, pour les méthodes de 3e génération, n’est pas suffisante. Cela étant, beaucoup d’autres génomes d’espèces plus exotiques ou appartenant à des territoires encore insuffisamment explorés, comme les océans, commencent à être identifiés et séquencés (expédition TARA),25 mais surtout beaucoup plus d’échantillons de chacune des espèces dont le génome est déjà connu seront analysés dans une quête du polymorphisme. Enfin la médecine, avec la perspective d’une approche personnalisée et d’une recherche des susceptibilités individuelles aux pathogènes, médicaments et désordres métaboliques, sera encore et pour longtemps demandeuse d’analyses.
Les laboratoires d’une petite poignée de pays dont principalement et de loin les États-Unis et le Royaume-Uni ont participé à cette aventure. En France, il faut bien le reconnaître, notre contribution a été modeste au regard de la taille du pays et de son importance dans le domaine scientifique. Malgré des débuts prometteurs, séquençage du génome du VHB, création du CEPH, création du Centre de recherche et d’étude des génomes (GREG), création par le CNRS du Génoscope qui séquença entre autres le chromosome 14 humain, la communauté scientifique du pays ne s’est pas, du moins au début, impliquée dans l’aventure. La raison de cette attitude doit être recherchée dans notre propension à privilégier la partie intellectuelle de la recherche trop souvent au détriment de la technique. Il est peu vraisemblable qu’un chercheur comme Frederick Sanger qui, pendant des années, a essayé de développer une méthode à partir d’un phage monocaténaire ayant au départ peu de chance d’être opérationnelle avec de l’ADN double brin aurait été soutenu financièrement, intellectuellement par son institution. Fort heureusement, devant l’importance prise ces dernières années par l’analyse de l’ADN, les esprits ont quelque peu évolué favorablement.
Il est toujours difficile de prédire l’avenir, toutefois on peut penser que les développements technologiques vont porter plus sur la recherche d’une baisse des coûts, d’une augmentation de la fiabilité des résultats qui, pour les méthodes de 3e génération, n’est pas suffisante. Cela étant, beaucoup d’autres génomes d’espèces plus exotiques ou appartenant à des territoires encore insuffisamment explorés, comme les océans, commencent à être identifiés et séquencés (expédition TARA),25 mais surtout beaucoup plus d’échantillons de chacune des espèces dont le génome est déjà connu seront analysés dans une quête du polymorphisme. Enfin la médecine, avec la perspective d’une approche personnalisée et d’une recherche des susceptibilités individuelles aux pathogènes, médicaments et désordres métaboliques, sera encore et pour longtemps demandeuse d’analyses.
* Le séquençage de l’ADN, 50 ans de prouesses technologiques et d’avancées scientifiques (Partie 1). Rev prat 2021;71:109-14.
Références
1. Sanger F, Nicklen S, Coulson AR.
DNA sequencing with chain-terminating inhibitors.
Proc Natl Acad Sci USA 1977;74:5463-7.
2. Gilbert, W, Maxam, A. The nucleotide sequence of the lac operator. Proc Natl Acad Sci USA 1973;70:3581-4.
3. Caruthers MH, Beaucage SL, Becker C, et al. Deoxyoligonucleotide synthesis via the phosphoramidite method. Gene Amplif Anal 1983;3:1-26.
4. Smith LM, Sanders JZ, Kaiser RJ et al., Fluorescence detection in automated DNA sequence analysis. Nature 1986;321:674-9.
5. Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 1986;51 Pt 1:263-73.
6. Staden R. A strategy of DNA sequencing employing computer programs. Nucleic Acid Res 1979;6:2601-10.
7. Sanger F, Coulson AR, Barrell BG, Smith AJ, Roe BA. Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing. J Mol Biol 1980;143:161-78.
8. Anderson S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res1981;9:3015-27.
9. Messing J, Gronenborn, B, Muller-Hill B, Hans Hofschneider P. FilamentouscoliphageM13 as a cloning vehicle: Insertion of a HindII fragment of the lac regulatory regionin M13 replicative form in vitro. Proc Natl Acad USA 1977;74:3642-6.
10. Dulbecco R. A turning point in cancer research: sequencing the human genome. Science1986;231:1055-6.
11. The C. elegans sequencing consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 1998;282:2012-8
12. Ewing B, Hillier L, Wendl MC, Green P. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Res 1998;8:175-85.
13. Green P. Phrap documentation, 1996 http://www.phrap.org/phredphrap/phrap.html
14. Gordon D, Abajian A, Green P. Consed : a graphical tool for sequence finishing. Genome Res 1998;8:195-202.
15. Lander ES, Lindon LM, Birren B, et al. Initial sequencing and analysis of the human genome Nature 2001;409:860-921.
16. Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995;269:496-512.
17. Adams MD, Celniker SE, Holt RA, et al. The genome sequence of drosophila melanogaster Science 2000;287:2185-95.
18. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science 2001;291:1304-51.
19. Richard E. Green RE, Krause J, et al. A draft sequence of the Neanderthal genome Science 2010;328:710-22.
20. Meyer M, Kircher M, Gansauge MT, et al. A high coverage genome sequence from an archaic Denisovan individual. Science 2012;338:222-6.
21. Ronaghi M, Karamohamed S, Pettersson B, Uhlén M, Nyrén P. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 1996;242:84-9.
22. https://www.illumina.com/systems/ sequencing-platforms.html
23. Eid J, Fehr A, Gray J, et al. Real time DNA sequencing from single polymerase molecules. Science 2009;323:133-8.
24. Milten J, Koren S, Rand AC, et al. Nanopore sequencing and assembly of a human genome with ultra-long read. Nature Biotechnology 2018;36:338-45.
25. Venter JC, Remington K, Heidelberg JF, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004;304:66-74.
2. Gilbert, W, Maxam, A. The nucleotide sequence of the lac operator. Proc Natl Acad Sci USA 1973;70:3581-4.
3. Caruthers MH, Beaucage SL, Becker C, et al. Deoxyoligonucleotide synthesis via the phosphoramidite method. Gene Amplif Anal 1983;3:1-26.
4. Smith LM, Sanders JZ, Kaiser RJ et al., Fluorescence detection in automated DNA sequence analysis. Nature 1986;321:674-9.
5. Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 1986;51 Pt 1:263-73.
6. Staden R. A strategy of DNA sequencing employing computer programs. Nucleic Acid Res 1979;6:2601-10.
7. Sanger F, Coulson AR, Barrell BG, Smith AJ, Roe BA. Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing. J Mol Biol 1980;143:161-78.
8. Anderson S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res1981;9:3015-27.
9. Messing J, Gronenborn, B, Muller-Hill B, Hans Hofschneider P. FilamentouscoliphageM13 as a cloning vehicle: Insertion of a HindII fragment of the lac regulatory regionin M13 replicative form in vitro. Proc Natl Acad USA 1977;74:3642-6.
10. Dulbecco R. A turning point in cancer research: sequencing the human genome. Science1986;231:1055-6.
11. The C. elegans sequencing consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 1998;282:2012-8
12. Ewing B, Hillier L, Wendl MC, Green P. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Res 1998;8:175-85.
13. Green P. Phrap documentation, 1996 http://www.phrap.org/phredphrap/phrap.html
14. Gordon D, Abajian A, Green P. Consed : a graphical tool for sequence finishing. Genome Res 1998;8:195-202.
15. Lander ES, Lindon LM, Birren B, et al. Initial sequencing and analysis of the human genome Nature 2001;409:860-921.
16. Fleischmann RD, Adams MD, White O, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995;269:496-512.
17. Adams MD, Celniker SE, Holt RA, et al. The genome sequence of drosophila melanogaster Science 2000;287:2185-95.
18. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science 2001;291:1304-51.
19. Richard E. Green RE, Krause J, et al. A draft sequence of the Neanderthal genome Science 2010;328:710-22.
20. Meyer M, Kircher M, Gansauge MT, et al. A high coverage genome sequence from an archaic Denisovan individual. Science 2012;338:222-6.
21. Ronaghi M, Karamohamed S, Pettersson B, Uhlén M, Nyrén P. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 1996;242:84-9.
22. https://www.illumina.com/systems/ sequencing-platforms.html
23. Eid J, Fehr A, Gray J, et al. Real time DNA sequencing from single polymerase molecules. Science 2009;323:133-8.
24. Milten J, Koren S, Rand AC, et al. Nanopore sequencing and assembly of a human genome with ultra-long read. Nature Biotechnology 2018;36:338-45.
25. Venter JC, Remington K, Heidelberg JF, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004;304:66-74.
Dans cet article
- D’autres avancées spectaculaires
- Le programme « génome humain » (1989)
- Le nématode C. elegans (1998)
- La mouche du vinaigre (2000)
- Le brouillon du génome humain (2001)
- Nouvelles méthodes de séquençage ou méthodes de 2e génération
- Méthodes de troisième génération
- Des développements majeurs dans de multiples domaines