La macroglobulinémie de Waldenström est un syndrome lymphoprolifératif B, rare et indolent, rattaché au groupe des lymphomes lymphoplasmocytaires de la classification établie en 2016 par l’Organisation mondiale de la santé.1
Les critères diagnostiques de cette maladie comportent une infiltration médullaire diffuse par des cellules lymphoplasmocytaires et la présence d’une immunoglobuline de type M (IgM) monoclonale sérique quelle qu’en soit la concentration.
Historiquement, cette hémopathie a été décrite pour la première fois en 1944 par Jan Gösta Waldenström. Ce médecin suédois a observé, chez deux patientes, l’association d’une épistaxis, de gingivorragies et d’adénopathies.
Les critères diagnostiques de cette maladie comportent une infiltration médullaire diffuse par des cellules lymphoplasmocytaires et la présence d’une immunoglobuline de type M (IgM) monoclonale sérique quelle qu’en soit la concentration.
Historiquement, cette hémopathie a été décrite pour la première fois en 1944 par Jan Gösta Waldenström. Ce médecin suédois a observé, chez deux patientes, l’association d’une épistaxis, de gingivorragies et d’adénopathies.
Épidémiologie
La maladie de Waldenström est une maladie rare représentant 1 à 2 % des hémopathies et 6 % des syndromes lymphoprolifératifs. Deux fois plus fréquente chez l’homme, son incidence est estimée à 3 nouveaux cas par million d’habitants. L’âge médian au moment du diag- nostic est de 70 ans et la survie médiane est de 8 ans à partir du premier traitement.2
Des facteurs de risque de développer une maladie de Waldenström ont été identifiés. Le principal étant la présence d’une gammapathie monoclonale de signification indéterminée (monoclonal gammapathy of undetermined significance [MGUS]) d’isotype IgM. Elle se caractérise par la présence d’une IgM monoclonale sans infiltration lymphomateuse médullaire. Elle est présente chez 1 personne sur 600 au-delà de 50 ans et multiplie par 46 le risque de développer une maladie de Waldenström avec un risque de progression de 10 % à 5 ans, et de 24 % à 15 ans.3 La détection d’une IgM monoclonale impose la réalisation d’une évaluation médullaire chez tous les patients ayant un symptôme évocateur de maladie de Waldenström. Chez les patients asymptomatiques, la nécessité d’un examen de la moelle osseuse est plus controversée (les patients asymptomatiques ne néces- sitant pas de traitement).
Il existe une prédisposition familiale puisque 20 % des patients atteints ont un parent de 1er degré avec un désordre lymphocytaire B.
Certaines pathologies auto-immunes sont plus fréquemment décrites dans les familles de patients ayant une maladie de Waldenström ; on peut citer la polyarthrite rhumatoïde, la maladie cœliaque et la sarcoïdose.
Des facteurs de risque de développer une maladie de Waldenström ont été identifiés. Le principal étant la présence d’une gammapathie monoclonale de signification indéterminée (monoclonal gammapathy of undetermined significance [MGUS]) d’isotype IgM. Elle se caractérise par la présence d’une IgM monoclonale sans infiltration lymphomateuse médullaire. Elle est présente chez 1 personne sur 600 au-delà de 50 ans et multiplie par 46 le risque de développer une maladie de Waldenström avec un risque de progression de 10 % à 5 ans, et de 24 % à 15 ans.3 La détection d’une IgM monoclonale impose la réalisation d’une évaluation médullaire chez tous les patients ayant un symptôme évocateur de maladie de Waldenström. Chez les patients asymptomatiques, la nécessité d’un examen de la moelle osseuse est plus controversée (les patients asymptomatiques ne néces- sitant pas de traitement).
Il existe une prédisposition familiale puisque 20 % des patients atteints ont un parent de 1er degré avec un désordre lymphocytaire B.
Certaines pathologies auto-immunes sont plus fréquemment décrites dans les familles de patients ayant une maladie de Waldenström ; on peut citer la polyarthrite rhumatoïde, la maladie cœliaque et la sarcoïdose.
Circonstances de découverte, manifestations cliniques et biologiques
Infiltration lymphomateuse
L’infiltration médullaire peut induire des cytopénies. L’anémie, fréquente dans la maladie de Waldenström, peut être multifactorielle (infiltration, hémodilution, inflammation), habituellement normocytaire et aré- générative. Un syndrome inflammatoire (augmentation de la protéine C-réactive) est en effet fréquent, pouvant entraîner une anémie significative et être un critère d’initiation de traitement. L’accélération de la vitesse de sédimentation est liée à l’immunoglobuline mono- clonale et ne traduit pas nécessairement un syndrome inflammatoire.
Le syndrome tumoral ganglionnaire-splénomégalie est moins fréquent que pour d’autres types de lymphomes (15-20 % lors du diagnostic).4 D’autres atteintes d’organes sont décrites (poumons, tube digestif, reins, peau, et système nerveux central [syndrome de Bing-Neel].
Le syndrome tumoral ganglionnaire-splénomégalie est moins fréquent que pour d’autres types de lymphomes (15-20 % lors du diagnostic).4 D’autres atteintes d’organes sont décrites (poumons, tube digestif, reins, peau, et système nerveux central [syndrome de Bing-Neel].
Propriétés physico-chimiques de l’IgM
L’IgM monoclonale donne une dimension unique à cette pathologie, du fait de ses propriétés physico-chimiques (haut poids moléculaire et capacité à polymériser en pentamère) et antigéniques.
Le syndrome d’hyperviscosité est observé lorsque le taux d’IgM plasmatique est élevé, en général à partir de 30 g/L (mais une importante variabilité interindividuelle est constatée). Présent chez 15 % des patients au diagnostic, ce syndrome peut occasionner des céphalées, une vision trouble, une épistaxis ou des vertiges. Le fond d’œil est un indicateur précoce d’hyperviscosité en montrant des signes d’hémorragie maculaire, et des veines tortueuses et dilatées. La prise en charge repose initialement sur la réalisation urgente de séances de plasma- phérèse en milieu spécialisé, permettant de soustraire les macromolécules IgM. Classiquement, une séance de plasmaphérèse réduit le taux d’IgM de 20 à 30 % et permet de traiter efficacement et rapidement un syndrome d’hyperviscosité. Les séances de plasmaphérèse sont ensuite suivies d’un traitement de chimiothérapie.
Le syndrome de Willebrand acquis se définit comme un syndrome hémorragique comportant les anomalies biologiques de la maladie de Willebrand constitutionnelle se développant en association avec une pathologie, ici la maladie de Waldenström. Il est souvent le reflet d’un syndrome d’hyperviscosité qui entraîne le cisaillement des facteurs de la coagulation.
L’amylose AL associée à une IgM monoclonale est retrouvée chez 2 % des patients ayant une gammapathie à IgM. L’atteinte ganglionnaire et pulmonaire y est plus fréquente.
La cryoglobulinémie de type I constituée par l’IgM monoclonale, qui a alors la capacité de précipiter au froid, entraîne une altération de la microcirculation avec obstruction vasculaire. Elle serait symptomatique dans 5 % des cas, en créant une vascularite systémique (phénomènes de Raynaud, acrocyanoses, arthralgies, purpura), une neuropathie périphérique, ou des lésions rénales.
Le syndrome d’hyperviscosité est observé lorsque le taux d’IgM plasmatique est élevé, en général à partir de 30 g/L (mais une importante variabilité interindividuelle est constatée). Présent chez 15 % des patients au diagnostic, ce syndrome peut occasionner des céphalées, une vision trouble, une épistaxis ou des vertiges. Le fond d’œil est un indicateur précoce d’hyperviscosité en montrant des signes d’hémorragie maculaire, et des veines tortueuses et dilatées. La prise en charge repose initialement sur la réalisation urgente de séances de plasma- phérèse en milieu spécialisé, permettant de soustraire les macromolécules IgM. Classiquement, une séance de plasmaphérèse réduit le taux d’IgM de 20 à 30 % et permet de traiter efficacement et rapidement un syndrome d’hyperviscosité. Les séances de plasmaphérèse sont ensuite suivies d’un traitement de chimiothérapie.
Le syndrome de Willebrand acquis se définit comme un syndrome hémorragique comportant les anomalies biologiques de la maladie de Willebrand constitutionnelle se développant en association avec une pathologie, ici la maladie de Waldenström. Il est souvent le reflet d’un syndrome d’hyperviscosité qui entraîne le cisaillement des facteurs de la coagulation.
L’amylose AL associée à une IgM monoclonale est retrouvée chez 2 % des patients ayant une gammapathie à IgM. L’atteinte ganglionnaire et pulmonaire y est plus fréquente.
La cryoglobulinémie de type I constituée par l’IgM monoclonale, qui a alors la capacité de précipiter au froid, entraîne une altération de la microcirculation avec obstruction vasculaire. Elle serait symptomatique dans 5 % des cas, en créant une vascularite systémique (phénomènes de Raynaud, acrocyanoses, arthralgies, purpura), une neuropathie périphérique, ou des lésions rénales.
Propriétés immunologiques de l’IgM
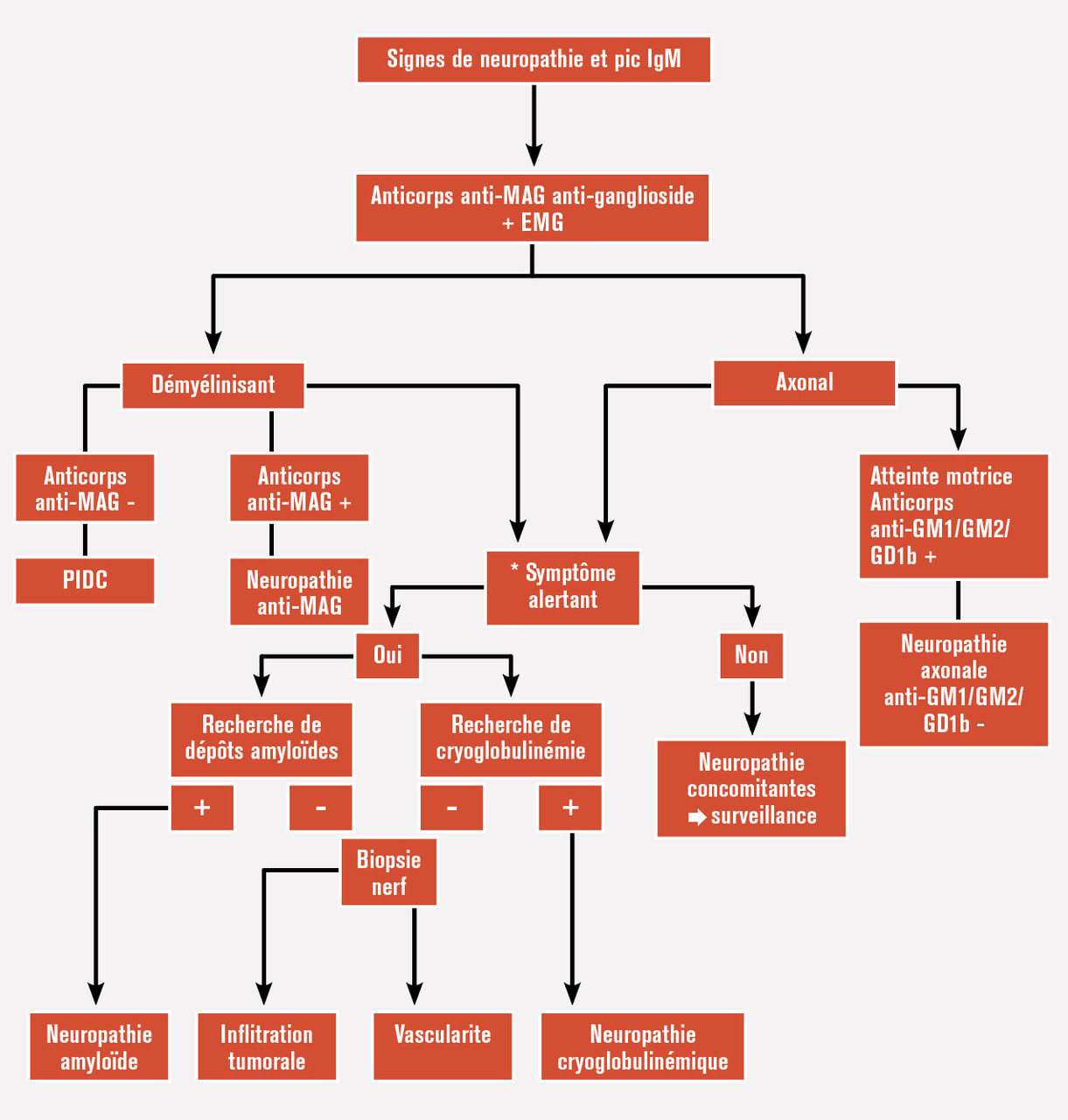
Une neuropathie périphérique est présente chez 25 % des patients (v. figure). La neuropathie la plus fréquente est secondaire à une activité anti-MAG (myelin-associated glycoprotein) de l’IgM monoclonale. L’atteinte est initialement sensitive, progressive, distale et symétrique. Elle débute avec des paresthésies prédominant aux membres inférieurs avec hypoesthésie en chaussette, une ataxie ou des tremblements posturaux. L’aréflexie est diffuse, sans atteinte des nerfs crâniens ou dysautonomie. L’évolution est lente, et un déficit moteur distal et symétrique (notamment atteinte des muscles releveurs des pieds) peut apparaître. D’autres anticorps ont été décrits (anticorps anti-gangliosides ou anti-sulfatides), mais l’absence de détection d’anticorps n’exclut pas le diagnostic de neuropathie périphérique associé à la maladie de Waldenström. Les tableaux cliniques sont hétérogènes et la démarche diagnostique souvent complexe.
L’association à des signes neurologiques centraux, la rapidité d’évolution ou l’aggravation récente d’une neuropathie périphérique doivent faire rechercher une atteinte centrale. La réalisation d’une ponction lombaire ainsi qu’une imagerie par résonance magnétique cérébrale et médullaire sont indiquées.
La cryoglobulinémie de type II ; l’IgM monoclonale peut avoir une activité facteur rhumatoïde en reconnaissant le fragment constant des IgG aboutissant à la formation de complexes immuns. On observe un dépôt de ces complexes immuns dans les vaisseaux de petit calibre, activant le complément et pouvant entraîner une vas- cularite systémique.
La maladie des agglutinines froides constitue un tableau d’anémie hémolytique auto-immune. Le tableau clinique, lors de l’exposition à des températures basses, peut comporter un syndrome de Raynaud, une acrocyanose ou un livedo reticularis. Elle est suspectée devant un tableau clinique et biologique d’anémie hémolytique auto-immune avec un test de Coombs direct positif de type complément (C3). Le diagnostic est porté par la mise en évidence d’agglutinines froides dans le sérum. De récentes études tendent à penser que ce tableau entre dans le cadre d’un syndrome lymphoprolifératif avec IgM monoclonale, distinct d’une maladie de Waldenström.5
Le syndrome de Schnitzler est défini par l’association d’une urticaire chronique, accompagnée de fièvre, d’arthralgies, de douleurs osseuses et d’adénopathies. Les mécanismes physiopathologiques demeurent inconnus, mais l’interleukine 1 (IL-1) semble jouer un rôle crucial dans cette présentation avec activation de processus inflammatoires.
L’association à des signes neurologiques centraux, la rapidité d’évolution ou l’aggravation récente d’une neuropathie périphérique doivent faire rechercher une atteinte centrale. La réalisation d’une ponction lombaire ainsi qu’une imagerie par résonance magnétique cérébrale et médullaire sont indiquées.
La cryoglobulinémie de type II ; l’IgM monoclonale peut avoir une activité facteur rhumatoïde en reconnaissant le fragment constant des IgG aboutissant à la formation de complexes immuns. On observe un dépôt de ces complexes immuns dans les vaisseaux de petit calibre, activant le complément et pouvant entraîner une vas- cularite systémique.
La maladie des agglutinines froides constitue un tableau d’anémie hémolytique auto-immune. Le tableau clinique, lors de l’exposition à des températures basses, peut comporter un syndrome de Raynaud, une acrocyanose ou un livedo reticularis. Elle est suspectée devant un tableau clinique et biologique d’anémie hémolytique auto-immune avec un test de Coombs direct positif de type complément (C3). Le diagnostic est porté par la mise en évidence d’agglutinines froides dans le sérum. De récentes études tendent à penser que ce tableau entre dans le cadre d’un syndrome lymphoprolifératif avec IgM monoclonale, distinct d’une maladie de Waldenström.5
Le syndrome de Schnitzler est défini par l’association d’une urticaire chronique, accompagnée de fièvre, d’arthralgies, de douleurs osseuses et d’adénopathies. Les mécanismes physiopathologiques demeurent inconnus, mais l’interleukine 1 (IL-1) semble jouer un rôle crucial dans cette présentation avec activation de processus inflammatoires.
Bilan diagnostique et d’extension
Le bilan biologique nécessaire est résumé dans le tableau 1.
Le diagnostic est effectué sur l’analyse médullaire, habituellement sur la biopsie ostéomédullaire. Le myé- logramme avec aspiration simple permet également de donner les informations nécessaires au diagnostic, à savoir une infiltration lymphoplasmocytaire. Un immunophénotypage lymphocytaire permet de carac- tériser cette population lymphoplasmocytaire et d’éli- miner certaines pathologies lymphoïdes comme un lymphome lymphocytique, un myélome IgM ou un lymphome folliculaire.
Une analyse cytogénétique est également effectuée, retrouvant une anomalie dans 45 à 50 % des cas, sans spécificité pour la maladie de Waldenström et sans caractère pronostique (hormis, récemment, la délétion 17p).6
L’analyse moléculaire comprend la recherche de la mutation du gène MYD88. En effet, celle-ci est présente chez 90 à 95 % des patients ayant une maladie de Waldenström.7 La principale mutation récurrente est la mutation somatique, gain de fonction, L265P sur le gène MYD88. La protéine MYD88 est une molécule adaptatrice, et intervient lors de l’activation de récepteurs Toll-like (TLR) et du récepteur de l’IL-1. La mutation L265P a pour conséquence une activation de la voie de signalisation NFκB, en impliquant la tyrosine kinase de Bruton (BTK) et les kinases associées au récepteur de l’IL-1 (IRAK1 et IRAK4) conduisant à la croissance et la survie cellulaire.
Cette mutation est rarement présente dans les autres types de lymphomes partageant des caractéristiques avec la maladie de Waldenström (comme le lymphome de la zone marginale), faisant de la recherche de cette mutation un outil diagnostique.
La présence de la mutation est associée à une survie plus longue, et son absence à une résistance à l’ibrutinib.
La mutation du gène CXCR4 est trouvée dans 30 à 40 % des cas, mais n’est pas encore recherchée en routine, car elle n’a pas de répercussion diagnostique. La protéine CXCR4 est un récepteur de chimiokines (CXCL12) ; son activation entraîne la survie, la migration et l’adhésion cellulaire.
Le diagnostic est effectué sur l’analyse médullaire, habituellement sur la biopsie ostéomédullaire. Le myé- logramme avec aspiration simple permet également de donner les informations nécessaires au diagnostic, à savoir une infiltration lymphoplasmocytaire. Un immunophénotypage lymphocytaire permet de carac- tériser cette population lymphoplasmocytaire et d’éli- miner certaines pathologies lymphoïdes comme un lymphome lymphocytique, un myélome IgM ou un lymphome folliculaire.
Une analyse cytogénétique est également effectuée, retrouvant une anomalie dans 45 à 50 % des cas, sans spécificité pour la maladie de Waldenström et sans caractère pronostique (hormis, récemment, la délétion 17p).6
L’analyse moléculaire comprend la recherche de la mutation du gène MYD88. En effet, celle-ci est présente chez 90 à 95 % des patients ayant une maladie de Waldenström.7 La principale mutation récurrente est la mutation somatique, gain de fonction, L265P sur le gène MYD88. La protéine MYD88 est une molécule adaptatrice, et intervient lors de l’activation de récepteurs Toll-like (TLR) et du récepteur de l’IL-1. La mutation L265P a pour conséquence une activation de la voie de signalisation NFκB, en impliquant la tyrosine kinase de Bruton (BTK) et les kinases associées au récepteur de l’IL-1 (IRAK1 et IRAK4) conduisant à la croissance et la survie cellulaire.
Cette mutation est rarement présente dans les autres types de lymphomes partageant des caractéristiques avec la maladie de Waldenström (comme le lymphome de la zone marginale), faisant de la recherche de cette mutation un outil diagnostique.
La présence de la mutation est associée à une survie plus longue, et son absence à une résistance à l’ibrutinib.
La mutation du gène CXCR4 est trouvée dans 30 à 40 % des cas, mais n’est pas encore recherchée en routine, car elle n’a pas de répercussion diagnostique. La protéine CXCR4 est un récepteur de chimiokines (CXCL12) ; son activation entraîne la survie, la migration et l’adhésion cellulaire.
Diagnostics différentiels
Le diagnostic différentiel principal est la gammapathie monoclonale de signification indéterminée à IgM. L’existence d’un pic monoclonal sérique de type IgM associé à une infiltration lymphoïde peut être observé dans différentes hémopathies lymphoïdes chroniques B telles que la leucémie lymphoïde chronique, les lymphomes à cellules du manteau, le lymphome folliculaire, les lymphomes de la zone marginale, ou le myélome à IgM. La cytologie médullaire et l’analyse histologique, les techniques d’immunophénotypage ainsi que la cytogénétique constituent des outils indispensables pour établir le diagnostic différentiel entre la maladie de Waldenström et les autres hémopathies lymphoïdes chroniques B. La présence d’un composant lymphoplasmocytaire sur l’histologie de la moelle osseuse reste un argument fortement en faveur de la maladie de Waldenström. Le diagnostic différentiel avec le lymphome de la zone marginale peut être le plus difficile.
La recherche de la mutation L265P du gène MYD88 apporte à présent une aide importante au diagnostic.
La recherche de la mutation L265P du gène MYD88 apporte à présent une aide importante au diagnostic.
Pronostic
Le score IPSS (international prognostic staging system) a été proposé pour les patients ayant une maladie de Waldenström symptomatique.8 L’analyse de 587 cas de maladie de Waldenström a permis d’identifier cinq facteurs impactant la survie (tableau 2) :8
– âge > 65 ans ;
– hémoglobine ≤ 11,5 g/dL ;
– plaquettes ≤ 100 000/mm3 ;
– bêta-2-microglobuline > 3 mg/L ;
– IgM monoclonale > 70 g/L.
Ces facteurs permettent de déterminer trois groupes pronostiques différents en terme de survie globale médiane.8
Récemment, le génotype MYD88 non muté associé à CXCR4 non muté a été associé à une survie globale inférieure et à une résistance à certains médicaments.
– âge > 65 ans ;
– hémoglobine ≤ 11,5 g/dL ;
– plaquettes ≤ 100 000/mm3 ;
– bêta-2-microglobuline > 3 mg/L ;
– IgM monoclonale > 70 g/L.
Ces facteurs permettent de déterminer trois groupes pronostiques différents en terme de survie globale médiane.8
Récemment, le génotype MYD88 non muté associé à CXCR4 non muté a été associé à une survie globale inférieure et à une résistance à certains médicaments.
Traitement
Indication
Un traitement est initié uniquement chez les patients ayant une maladie symptomatique (liée à l’IgM ou au syndrome tumoral) [tableau 3].9 Le taux d’IgM, pris isolément, ne constitue pas en soi une indication thérapeutique. En effet, il n’a jamais été démontré de bénéfice au traitement d’une maladie de Waldenström asymp- tomatique. Celle-ci peut rester stable de nombreuses années. La surveillance est donc préconisée chez ces patients. Au moment du diagnostic, 30 à 50 % des patients sont asymptomatiques et ne nécessitent pas de traitement. Le risque de progression à 5 ans est de 59 %.
La présence d’une anémie est l’indication de traitement la plus fréquemment retrouvée.
La présence d’une anémie est l’indication de traitement la plus fréquemment retrouvée.
Options thérapeutiques
La difficulté dans le choix entre les différentes options thérapeutiques réside dans le manque d’essais prospectifs randomisés. Les traitements ont longtemps reposé sur l’utilisation des agents alkylants (chloraminophène, cyclophosphamide) et des analogues des purines (fludarabine). Bien que ces agents soient toujours utilisés, d’importants progrès ont cependant été réalisés avec l’introduction des anticorps monoclonaux anti-CD20 (rituximab), l’utilisation de la bendamustine ou des inhibiteurs du protéasome (bortézomib) et, plus récemment, l’apparition de thérapies ciblées (ibrutinib, idélalisib).
Parmi toutes les options thérapeutiques, le choix se fait principalement en fonction de la nécessité ou non d’obtenir une réponse rapide et en fonction du profil de toxicité des différents agents thérapeutiques.
Malgré ces importants progrès, peu de patients atteignent une rémission complète. Au-delà de l’augmentation de la survie, l’amélioration de la qualité de vie est donc un objectif principal.
L’association rituximab, cyclophosphamide et dexaméthasone est actuellement considérée comme un traitement standard de première ligne.10
L’utilisation du rituximab en monothérapie montre des taux de réponse plutôt bas (50 %) avec une survie globale médiane de 85 % à 5 ans. La tolérance est acceptable quel que soit l’âge du patient ou les facteurs de comorbidités. Le rituximab en monothérapie est utilisé uniquement pour des patients fragiles avec de multiples comorbidités ou dans le cadre d’une symptomatologie en lien avec l’IgM (neuropathies, cryoglubulinémie). La réponse est lente (en médiane 6 mois). On peut observer des élévations transitoires du taux d’IgM au début du traitement par rituximab (effet flare) rapportées chez 50 % des patients, ne préjugeant pas de l’efficacité du traitement. L’effet flare peut provoquer une majoration de l’hyperviscosité, de même qu’une aggravation d’une neuropathie ou d’une cryoglobulinémie lié à l’IgM. La première perfusion de rituximab peut donc être différée à la deuxième cure.11
L’ibrutinib, inhibiteur de la tyrosine kinase de Bruton, a été évalué en monothérapie à la posologie de 420 mg/j, par voie orale. Les taux de réponse globale dépassent les 90 %.12 Les meilleurs taux de réponse sont observés chez les patients ayant la mutation L265P du gène MYD88 en l’absence de mutation du gène CXCR4. L’ibrutinib dispose actuellement d’une autorisation de mise sur le marché, en première ligne de traitement, aux États-Unis et en Europe, pour les patients ne pouvant avoir de chimiothérapie ou en rechute.
L’autogreffe de cellules souches hématopoïétiques est une option thérapeutique envisageable chez des patients jeunes, en situation de rechute précoce, avec des facteurs pronostiques défavorables. Compte tenu d’une morbi-mortalité non négligeable, la place de l’allogreffe ne fait pas l’objet d’un consensus à l’heure actuelle.
D’autres agents sont en cours d’essai, comme le vénétoclax, un inhibiteur de bcl2, induisant une apoptose cellulaire. Des thérapies ciblant CXCR4 ou MYD88 sont également en cours de développement.
Parmi toutes les options thérapeutiques, le choix se fait principalement en fonction de la nécessité ou non d’obtenir une réponse rapide et en fonction du profil de toxicité des différents agents thérapeutiques.
Malgré ces importants progrès, peu de patients atteignent une rémission complète. Au-delà de l’augmentation de la survie, l’amélioration de la qualité de vie est donc un objectif principal.
L’association rituximab, cyclophosphamide et dexaméthasone est actuellement considérée comme un traitement standard de première ligne.10
L’utilisation du rituximab en monothérapie montre des taux de réponse plutôt bas (50 %) avec une survie globale médiane de 85 % à 5 ans. La tolérance est acceptable quel que soit l’âge du patient ou les facteurs de comorbidités. Le rituximab en monothérapie est utilisé uniquement pour des patients fragiles avec de multiples comorbidités ou dans le cadre d’une symptomatologie en lien avec l’IgM (neuropathies, cryoglubulinémie). La réponse est lente (en médiane 6 mois). On peut observer des élévations transitoires du taux d’IgM au début du traitement par rituximab (effet flare) rapportées chez 50 % des patients, ne préjugeant pas de l’efficacité du traitement. L’effet flare peut provoquer une majoration de l’hyperviscosité, de même qu’une aggravation d’une neuropathie ou d’une cryoglobulinémie lié à l’IgM. La première perfusion de rituximab peut donc être différée à la deuxième cure.11
L’ibrutinib, inhibiteur de la tyrosine kinase de Bruton, a été évalué en monothérapie à la posologie de 420 mg/j, par voie orale. Les taux de réponse globale dépassent les 90 %.12 Les meilleurs taux de réponse sont observés chez les patients ayant la mutation L265P du gène MYD88 en l’absence de mutation du gène CXCR4. L’ibrutinib dispose actuellement d’une autorisation de mise sur le marché, en première ligne de traitement, aux États-Unis et en Europe, pour les patients ne pouvant avoir de chimiothérapie ou en rechute.
L’autogreffe de cellules souches hématopoïétiques est une option thérapeutique envisageable chez des patients jeunes, en situation de rechute précoce, avec des facteurs pronostiques défavorables. Compte tenu d’une morbi-mortalité non négligeable, la place de l’allogreffe ne fait pas l’objet d’un consensus à l’heure actuelle.
D’autres agents sont en cours d’essai, comme le vénétoclax, un inhibiteur de bcl2, induisant une apoptose cellulaire. Des thérapies ciblant CXCR4 ou MYD88 sont également en cours de développement.
UN TRAITEMENT BIENTÔT INFLUENCÉ PAR LA GÉNOMIQUE
En conclusion, les patients atteints de la maladie de Waldenström bénéficient d’approches thérapeutiques dont la tolérance est toujours plus acceptable. À l’avenir, le choix thérapeutique sera probablement influencé par la génomique. Les coûts économiques de ces nouvelles approches devront être examinés avec soin.
Références
1. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375-90.
2. Castillo JJ, Olszewski AJ, Kanan S, Meid K, Hunter ZR, Treon SP. Overall survival and competing risks of death in patients with Waldenström macroglobulinaemia: an analysis of the Surveillance, Epidemiology and End Results database. Br J Haematol 2015;169:81-9.
3. Kyle RA, Therneau TM, Rajkumar SV, et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood 2003;102:3759-64.
4. Fonseca R, Hayman S. Waldenström macroglobulinaemia. Br J Haematol 2007;138:700-20.
5. Randen U, Trøen G, Tierens A, et al. Primary cold agglutinin-associated lymphoproliferative disease: a B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica 2014;99:497-504.
6. Nguyen-Khac F, Lambert J, Chapiro E, et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenström’s macroglobulinemia. Haematologica 2013;98:649-54.
7. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012;367:826-33.
8. Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood 2009;113:4163-70.
9. Dimopoulos MA, Gertz MA, Kastritis E, et al. Update on treatment recommendations from the Fourth International Workshop on Waldenstrom’s Macroglobulinemia. J Clin Oncol 2009;27:120-6.
10. Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, et al. Primary treatment of Waldenström macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol 2007;25:3344-9.
11. Ghobrial IM, Fonseca R, Greipp PR, et al. Initial immunoglobulin M “flare” after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia: an Eastern Cooperative Oncology Group Study. Cancer 2004;101:2593-8.
12. Castillo JJ, Palomba ML, Advani R, Treon SP. Ibrutinib in Waldenström macroglobulinemia: latest evidence and clinical experience. Ther Adv Hematol 2016;7:179.
2. Castillo JJ, Olszewski AJ, Kanan S, Meid K, Hunter ZR, Treon SP. Overall survival and competing risks of death in patients with Waldenström macroglobulinaemia: an analysis of the Surveillance, Epidemiology and End Results database. Br J Haematol 2015;169:81-9.
3. Kyle RA, Therneau TM, Rajkumar SV, et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood 2003;102:3759-64.
4. Fonseca R, Hayman S. Waldenström macroglobulinaemia. Br J Haematol 2007;138:700-20.
5. Randen U, Trøen G, Tierens A, et al. Primary cold agglutinin-associated lymphoproliferative disease: a B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica 2014;99:497-504.
6. Nguyen-Khac F, Lambert J, Chapiro E, et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenström’s macroglobulinemia. Haematologica 2013;98:649-54.
7. Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012;367:826-33.
8. Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood 2009;113:4163-70.
9. Dimopoulos MA, Gertz MA, Kastritis E, et al. Update on treatment recommendations from the Fourth International Workshop on Waldenstrom’s Macroglobulinemia. J Clin Oncol 2009;27:120-6.
10. Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, et al. Primary treatment of Waldenström macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol 2007;25:3344-9.
11. Ghobrial IM, Fonseca R, Greipp PR, et al. Initial immunoglobulin M “flare” after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia: an Eastern Cooperative Oncology Group Study. Cancer 2004;101:2593-8.
12. Castillo JJ, Palomba ML, Advani R, Treon SP. Ibrutinib in Waldenström macroglobulinemia: latest evidence and clinical experience. Ther Adv Hematol 2016;7:179.
Dans cet article


Résumé
La maladie de Waldenström est un syndrome lymphoprolifératif B rare et indolent, représentant 1 à 2 % des hémopathies. Le spectre de ses complications est large ; elles sont liées à l’infiltration tumorale et aux caractéristiques physico-chimiques ou auto-immunes de l’IgM monoclonale. Une mutation somatique sur le gène MYD88 a été décrite dans la majorité des cas de maladie de Waldenström. Un traitement est nécessaire lorsque la maladie est symptomatique. Le choix thérapeutique dépend de l’âge du patient, des comorbidités et de la nécessité d’obtenir une réponse rapide. En première ligne, la majorité des patients sont traités avec une combinaison d’immunochimiothérapie de type rituximab-cyclophosphamide-dexaméthasone. Néanmoins, de nombreux nouveaux médicaments sont disponibles ou en développement. L’ibrutinib, inhibiteur de tyrosine kinase de Bruton, fait partie de l’arsenal thérapeutique avec une bonne efficacité thérapeutique dans la maladie de Waldenström, en première ligne et en rechute.