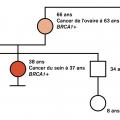
Imaginons que, dans le cadre de votre pratique, l’une de vos patientes, âgée de 43 ans, vous annonce qu’elle vient de subir une annexectomie totale de réduction de risque. Vous l’interrogez sur les raisons de cette chirurgie alors qu’elle ne présentait aucun problème gynécologique. Elle vous informe alors être porteuse asymptomatique d’un variant pathogène (également appelé mutation) de BRCA1, gène de prédisposition au cancer du sein et de l’ovaire. Sa sœur est en rémission d’un cancer du sein diagnostiqué alors qu’elle avait 37 ans, et sa mère a récemment développé un cancer de l’ovaire à 63 ans (
Apprenant par sa mère l’existence de ce variant pathogène, la patiente a pris rendez-vous auprès d’une équipe spécialisée d’oncogénétique. Lors de la consultation, après conseil génétique et recueil de son consentement écrit, un prélèvement sanguin a été réalisé pour un diagnostic présymptomatique. Le résultat est revenu positif. Sa sœur a entrepris la même démarche et elle est également porteuse.
BRCA1 et BRCA2, principaux gènes de prédisposition au cancer de l’ovaire
De 10 à 15 % des cancers épithéliaux de l’ovaire sont d’origine héréditaire, c’est-à-dire qu’ils sont associés à un variant pathogène constitutionnel dans un gène de prédisposition. Les principaux gènes de prédisposition au cancer de l’ovaire sont BRCA1 et BRCA2. Il s’agit de gènes suppresseurs de tumeurs codant pour des protéines impliquées dans la réparation de l’ADN, plus précisément dans les voies de recombinaison homologue. BRCA1 et BRCA2 assurent donc la stabilité du génome et ont ainsi un effet protecteur contre les cancers.
Les porteuses d’un variant pathogène ont un risque nettement augmenté de cancer ovarien, avec une incidence cumulée entre les âges de 35 et 80 ans de l’ordre de 44 % pour BRCA1 et de 17 % pour BRCA2, versus environ 1,5 % dans la population générale (
Les gènes BRCA1 et BRCA2 sont situés sur les autosomes, et non au niveau des chromosomes sexuels. Un homme peut ainsi être porteur d’un variant pathogène, tout autant qu’une femme, avec des risques de cancers cependant bien moindres (risque de cancer pancréatique déjà mentionné, risque de cancer du sein masculin [inférieur à 5 %] et risque de cancer de la prostate limité aux variants pathogènes de BRCA2).
Une patiente apprenant qu’elle est porteuse d’un variant pathogène a l’obligation légale d’en informer sa famille, pour que les proches qui le souhaitent puissent à leur tour se faire tester dans le cadre d’une consultation d’oncogénétique. On parle alors de « diagnostic (ou test) présymptomatique ». La transmission des variants pathogènes BRCA1/BRCA2 est dominante, ce qui signifie que chaque apparenté au premier degré (fratrie, parents, enfants) d’une personne porteuse a 50 % de risque de l’être également. Seules les personnes majeures peuvent bénéficier d’un test présymptomatique, en l’absence d’intérêt médical chez les enfants et adolescents.
Quelles implications pour la porteuse ?
La découverte d’un variant pathogène BRCA1 ou BRCA2 a plusieurs conséquences sur la prise en charge de la porteuse.
Pour la prise en charge ovarienne, chez une femme indemne, en l’absence de méthodes de dépistage efficaces, on recommande une annexectomie de réduction de risque. Il ne suffit donc pas de retirer les ovaires, il est impératif de retirer également les trompes de Fallope puisqu’une proportion des cancers séreux ovariens est d’origine tubaire. Les âges recommandés sont de 40 ans pour les porteuses du BRCA1, et de 45 ans pour les porteuses du BRCA2, avec un suivi clinique gynécologique dans les années qui précèdent la chirurgie. Sous réserve de l’absence d’antécédent personnel de cancer du sein, il est possible d’administrer à la patiente un traitement hormonal de substitution entre la chirurgie et l’âge naturel de la ménopause. Si l’annexectomie réduit considérablement le risque de cancer ovarien, il persiste néanmoins un faible risque résiduel dû aux dépôts péritonéaux de cellules d’origine ovarienne.
L’augmentation de risque de cancer du sein justifie un dépistage renforcé dès l’âge de 25 ans : réalisation d’une IRM annuelle, associée à celle d’une mammographie à partir de l’âge de 30 ans. Une mastectomie bilatérale de réduction de risque est envisageable chez une femme bien informée et qui en serait demandeuse.
La découverte des variants pathogènes BRCA1/BRCA2 a aussi désormais un intérêt théranostique pour les patientes en cours de traitement pour un cancer de l’ovaire ou du sein. En effet, sous certaines conditions, elles sont éligibles à un traitement par inhibiteur de poly-(ADP-ribose) polymérase (PARP) ciblant spécifiquement la déficience BRCA dans la cellule cancéreuse, en complément des chimiothérapies classiques.
Les autres gènes de prédisposition sont plus rarement impliqués
RAD51C, RAD51D, et PALB2 sont d’autres gènes de prédisposition au cancer de l’ovaire.3, 4 Comme BRCA1 et BRCA2, ils interviennent dans les systèmes de réparation de l’ADN. Les variants pathogènes de ces gènes sont plus rarement impliqués que ceux de BRCA1 et BRCA2. Le risque cumulé de cancer ovarien est de l’ordre de 10 % pour RAD51C et RAD51D, avec recommandation d’annexectomie de réduction de risque à la ménopause, et de 3 à 5 % pour PALB2 (
Enfin, des cancers ovariens sont aussi observés dans le syndrome de Lynch, syndrome de prédisposition principalement au cancer du côlon et de l’endomètre avec, pour leur part, une nette prédominance des types histologiques endométrioïdes et à cellules claires.6 Les gènes impliqués sont ceux du système de réparation de mésappariement de base (MMR) MLH1, MLS2, MSH6 et PMS2. Dans ce syndrome, la chirurgie gynécologique de réduction de risque est surtout justifiée par le risque de cancer endométrial : hystérectomie totale non conservatrice vers 40-45 ans.7
Un suivi régulier s’impose, même après une annexectomie
En cas de variant pathogène BRCA1, les patientes sont orientées vers un centre de suivi destiné aux femmes à risque génétique de cancer du sein et de l’ovaire par l’équipe d’oncogénétique qui les prend en charge.
Après une annexectomie de réduction de risque, le rôle du médecin traitant est de s’assurer que la patiente réalise son dépistage annuel du cancer du sein par IRM et mammographie, et qu’elle est avisée de la possibilité d’une mastectomie bilatérale de réduction de risque. Enfin, il est essentiel de lui rappeler l’obligation d’informer ses enfants de l’existence du variant pathogène BRCA1 dans la famille, afin qu’ils puissent à leur tour se faire tester lorsqu’ils seront jeunes adultes.
1. Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom MJ, et al. Risks of Breast, Ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017 Jun 20;317(23):2402-2416.
2. Li S, Silvestri V, Leslie G, Rebbeck TR, Neuhausen SL, Hopper JL, et al. Cancer risks associated with BRCA1 and BRCA2 pathogenic variants. J Clin Oncol 2022 May 10;40(14):1529-1541.
3. Song H, Dicks EM, Tyrer J, Intermaggio M, Chenevix-Trench G, Bowtell DD, et al. Population-based targeted sequencing of 54 candidate genes identifies PALB2 as a susceptibility gene for high-grade serous ovarian cancer. J Med Genet 2021;58(5):305‑13.
4. Yang X, Song H, Leslie G, Engel C, Hahnen E, Auber B, et al. Ovarian and breast cancer risks associated with pathogenic variants in RAD51C and RAD51D. JNCI J Natl Cancer Inst 2020;112(12):1242‑50.
5. Yang X, Leslie G, Doroszuk A, Schneider S, Allen J, Decker B, et al. Cancer risks associated with germline PALB2 pathogenic variants: An international study of 524 families. J Clin Oncol 2020;38(7):674‑85.
6. Crosbie EJ, Ryan NAJ, McVey RJ, Lalloo F, Bowers N, Green K, et al. Assessment of mismatch repair deficiency in ovarian cancer. J Med Genet 2021;58(10):687‑91.
7. Dominguez-Valentin M, Crosbie EJ, Engel C, Aretz S, Macrae F, Winship I, et al. Risk-reducing hysterectomy and bilateral salpingo-oophorectomy in female heterozygotes of pathogenic mismatch repair variants: a prospective Lynch syndrome database report. Genet Med 2021;23(4):705‑12.