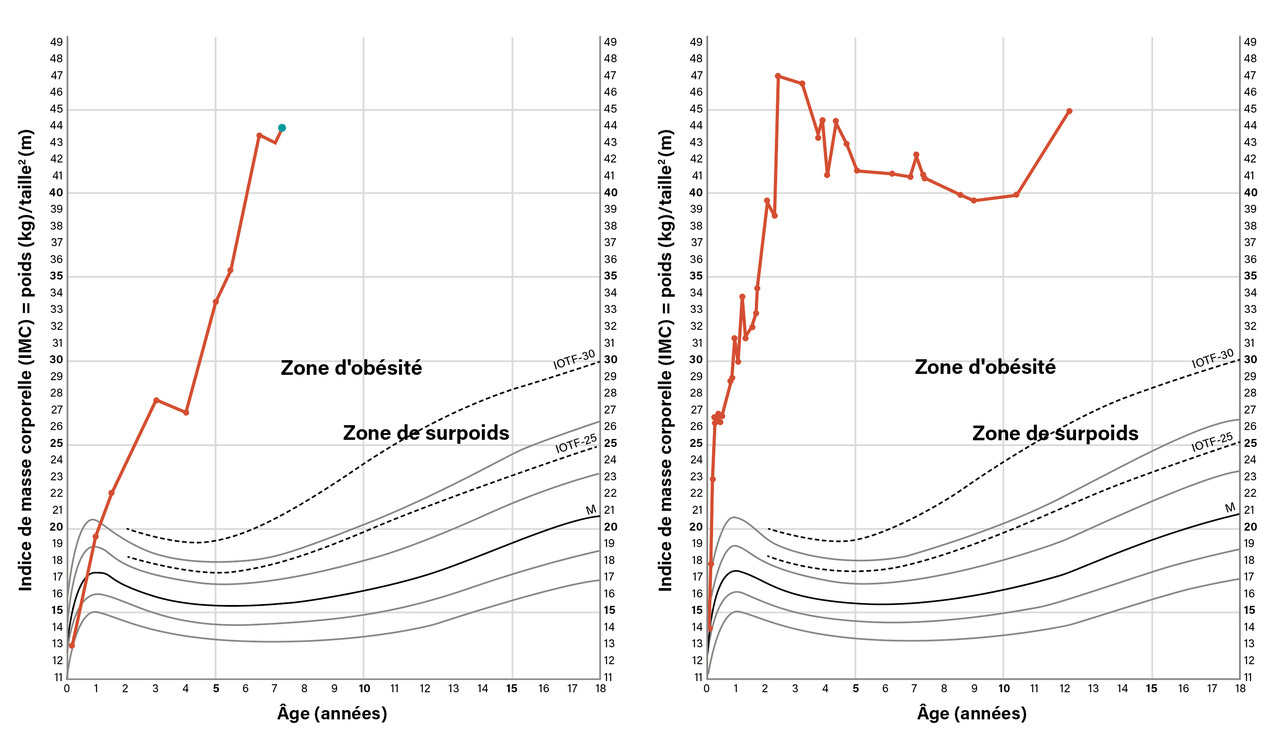
L’obésité, définie par un excès de masse grasse, résulte d’un déséquilibre de la balance énergétique. Son développement précoce au cours de la vie implique toujours une prédisposition génétique, en interaction avec des facteurs environnementaux multiples selon les situations (mode de vie, manque de sommeil, etc.).1, 2 Il s’agit donc d’une véritable maladie des centres de régulation de la faim dont l’héritabilité génétique peut atteindre jusqu’à 90 % dans les situations d’obésité sévère et précoce (débutant dès les premières années de vie).3
Les tableaux cliniques sont classiquement répartis en deux groupes : l’obésité dite polygénique, plus commune et plus fréquente (95 % des cas), et les obésités génétiques de causes rares ou obésités précoces et sévères associées ou non à un trouble du neurodéveloppement représentant au moins 5 % des cas.1, 4
Il est indispensable d’évoquer précocement un diagnostic d’obésité génétique rare afin de pouvoir proposer une prise en charge optimale adaptée le plus tôt possible et, le cas échéant, des traitements médicamenteux ciblés.5
La dysrégulation du contrôle hypothalamique de la faim et du poids est au centre du tableau clinique des obésités génétiques rares du fait de la réponse anormale aux signaux hormonaux venant de la périphérie (leptine, insuline, ghréline) et de l’altération des signaux venant du système nerveux autonome ou des voies hormonales hypophysaires.6 Une des voies clés altérée dans l’hypothalamus est la voie leptine-mélanocortines dont l’interruption entraîne une hyperphagie insatiable dès la petite enfance qui est particulièrement sévère pendant la phase de constitution de l’obésité. L’atteinte hypothalamique est aussi responsable d’une altération du métabolisme de base qui participe au développement anormal du tissu adipeux.
Deux catégories d’obésités génétiques rares
Il est classique de distinguer deux grandes catégories d’obésités génétiques rares : les obésités syndromiques et les obésités monogéniques non syndromiques.7, 8
Obésités syndromiques
Elles sont définies par une obésité associée à d’autres signes évocateurs de trouble neurodéveloppemental (déficience intellectuelle ou retard psychomoteur et/ou des apprentissages, ou troubles du spectre de l’autisme) et/ou d’un syndrome malformatif congénital (éléments dysmorphiques, anomalies d’organe). Les syndromes de Prader-Willi (SPW), de l’X fragile et de Bardet-Biedl (BBS) sont les plus fréquents.9, 10
Obésités monogéniques non syndromiques
Elles sont secondaires à un variant pathogène dans un gène codant pour une des protéines de la voie leptine-mélanocortines. Actuellement, plus de soixante gènes ont été décrits comme impliqués dans ces tableaux d’obésité monogénique.10 Les plus connus sont les gènes de la leptine (LEP), de son récepteur (LEPR), de la proopiomélanocortine (POMC), de la prohormone convertase subtilisine/kexine de type 1 (PCSK1), et du récepteur aux mélanocortines de type 4 (MC4R). Il s’agit d’obésités très précoces avec une hyperphagie majeure associées le plus souvent à des déficits endocriniens centraux (déficit corticotrope dans le cas du déficit en POMC ; hypogonadisme hypogonadotrope en cas de déficit en leptine ; diabète insipide en cas de déficit en PCSK1 par exemple).9, 11
Devant quel tableau clinique évoquer une obésité génétique ?
Toutes les obésités génétiques rares, qu’elles soient syndromiques ou monogéniques, ont une origine physiopathologique commune par l’atteinte des centres régulateurs hypothalamiques avec un véritable continuum entre les situations cliniques et un phénotype commun évocateur qui doit être repéré afin d’adapter la prise en charge le plus précocement possible. Le Programme national de diagnostic et de soins (PNDS) des obésités de causes rares, publié en juillet 2021 sur le site de la Haute Autorité de santé,12 oriente les cliniciens dans ces situations spécifiques.
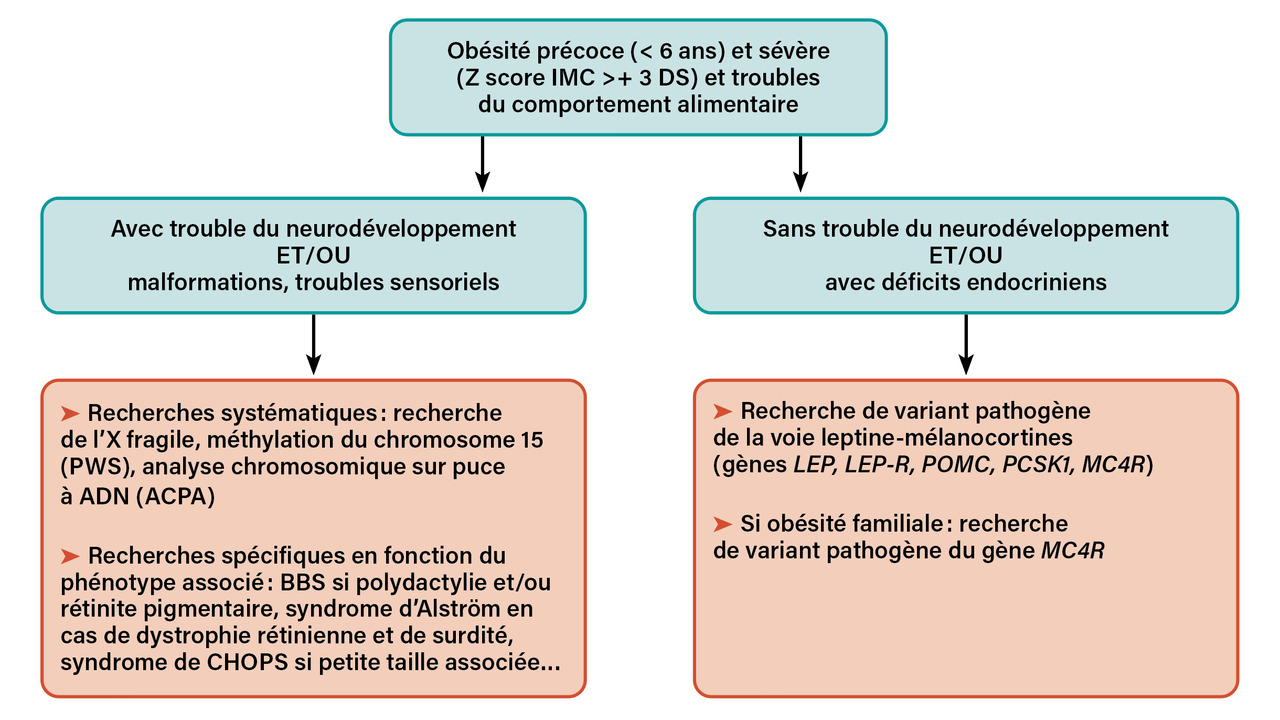
Les signes cliniques devant faire suspecter une obésité génétique (
D’autres atteintes sont associées de façon inconstante selon les causes :
– troubles endocriniens d’origine centrale (insuffisance corticotrope, puberté précoce ou retardée, hypothyroïdie centrale, déficit somatotrope entre autres) qui sont à rechercher en particulier en cas d’hypoglycémies néonatales, de retard statural ou de développement pubertaire anormal ;
– troubles du neurodéveloppement à rechercher à l’interrogatoire et à l’examen clinique (hypotonie néonatale, retard de développement psychomoteur et notamment retard de langage, déficience intellectuelle avec scolarité adaptée, troubles du spectre autistique et/ou troubles spécifiques des apprentissages).
La présence de malformations congénitales (polydactylie, malformation rénale ou cardiaque), d’atteintes neurosensorielles (ophtalmologiques comme un nystagmus ou une atteinte rétinienne, auditives) ou de particularités morphologiques associées à l’obésité est aussi évocatrice d’une origine génétique.5, 9
L’outil informatique ObsGen (http://obsgen.nutriomics.org), d’accès libre, développé par l'équipe INSERM Nutriomics U1269 (Prs Poitou et Dubern), permet d’orienter les cliniciens. Si besoin, les centres de référence et de compétence PRADORT (syndrome de Prader-Willi et autres obésités rares avec troubles du comportement alimentaire) peuvent aussi être contactés afin de discuter des situations particulières et aider à l’orientation étiologique.
Quels examens faire en cas de suspicion d’obésité génétique ?
Les analyses génétiques peuvent être discutées avec des généticiens cliniciens en lien avec un centre de référence des maladies rares (CRMR) de la filière DéfiScience. Elles dépendent du phénotype associé à l’obésité (
Des recherches plus spécifiques sont discutées dans un second temps, au cas par cas, avec le neurogénéticien en fonction du phénotype clinique. Les analyses plus larges type exome, voire génome, peuvent être indiquées dans certaines situations cliniques.7
Consultation génétique pour une prise en charge spécifique
En cas de diagnostic génétique établi, il est nécessaire d’orienter la famille vers une consultation génétique pour un conseil génétique et mettre en place une prise en charge adaptée et la plus spécifique possible comme discuté dans le Protocole national de diagnostic et de soins (PNDS) générique des obésités de causes rares.12 ●
1. Bouchard C. Genetics of obesity: What we have learned over decades of research. Obes Silver Spring Md 2021;29:802‑20.
2. Loos RJF, Janssens ACJW. Predicting polygenic obesity using genetic information. Cell Metab 2017;25:535‑43.
3. Silventoinen K, Jelenkovic A, Sund R, Hur YM, Yokoyama Y, Honda C, et al. Genetic and environmental effects on body mass index from infancy to the onset of adulthood: An individual-based pooled analysis of 45 twin cohorts participating in the COllaborative project of Development of Anthropometrical measures in Twins (CODATwins) study. Am J Clin Nutr 2016;104:371‑9.
4. Khera AV, Chaffin M, Wade KH, Zahid S, Brancale J, Xia R, et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 2019;177:587-596.e9.
5. Dubern B, Mosbah H, Pigeyre M, Clément K, Poitou C. Rare genetic causes of obesity: Diagnosis and management in clinical care. Ann Endocrinol 2022;83:63‑72.
6. Farooqi IS. Monogenic obesity syndromes provide insights into the hypothalamic regulation of appetite and associated behaviors. Biol Psychiatry 2022;9:856‑9.
7. Dubern B, Mosbah H, Pigeyre M, Clement K, Poitou C. Rare genetic causes of obesity: Diagnosis and management in clinical care. Ann Endocrinol 2021;S0003-4266(21)01109-4.
8. Huvenne H, Dubern B, Clement K, Poitou C. Rare genetic forms of obesity: Clinical Approach and Current Treatments in 2016. Obes Facts 2016;9:158‑73.
9. Huvenne H, Dubern B, Clement K, Poitou C. Rare Genetic Forms of Obesity: Clinical approach and current treatments in 2016. Obes Facts 2016;9:158‑73.
10. Pigeyre M, Yazdi FT, Kaur Y, Meyre D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin Sci 2016;130:943‑86.
11. Farooqi IS. Monogenic human obesity syndromes. Handb Clin Neurol 2021;181:301‑10.
12. Protocole national de diagnostic et de soins (PNDS). Générique Obésités de causes rares. HAS. 19 juillet 2021. https://vu.fr/jEbs
13. Huvenne H, Le Beyec J, Pepin D, Alili R, Pigeon Kherchiche P, Jeannic E, et al. Seven novel deleterious LEPR mutations found in early-onset obesity: A ΔExon6-8 shared by subjects from Reunion Island, France, suggests a founder effect. J Clin Endocrinol Metab 2015;100:E757-766.
14. Wabitsch M, Farooqi S, Flück CE, Bratina N, Mallya UG, Stewart M et al. Natural history of obesity due to POMC, PCSK1, and LEPR deficiency and the impact of setmelanotide. J Endocr Soc 2022;6:bvac057.