Les deux piliers du traitement de première ligne du lymphome de Hodgkin restent, en France en 2023, la polychimiothérapie associée à une radiothérapie de clôture dans les formes localisées (classification d’Ann Arbor, stades I-II) :1 le protocole ABVD (adriamycine, bléomycine, vinblastine et dacarbazine) est recommandé, associé, en fin de traitement, à une radiothérapie de clôture pour traiter les sites initialement atteints par la maladie. Ce traitement permet d’obtenir une rémission complète et durable chez 90 % des patients.2,3
Dans les formes étendues (classification d’Ann Arbor, stades III et IV), une polychimiothérapie plus intensive par BEACOPP escaladé (bléomycine, étoposide, doxorubicine, cyclophosphamide, vincristine, procarbazine et prednisone) est recommandée chez les patients jeunes (moins de 60 ans).4 Ce traitement permet d’obtenir une rémission complète et durable chez 85 % des patients. Si une rémission complète métabolique est obtenue après deux cycles de BEACOPP escaladé, une désescalade thérapeutique vers l’ABVD est recommandée, selon les données de l’étude thérapeutique AHL 2011.4
Le traitement de la rechute repose sur une polychimiothérapie de rattrapage intensif, avec une intensification thérapeutique réalisée sous couvert d’une autogreffe de cellules souches, chez les patients éligibles à ce traitement (moins de 60 ans).2,3
En situation de rechute, des traitements ciblés (anti-CD30, brentuximab vedotin) et les immunothérapies anti-PD1 (nivolumab ou pembrolizumab) peuvent être proposés.
Les profils de tolérance du brentuximab vedotin et des anti-PD1, et la gestion des principaux effets indésirables, dans le contexte du traitement du lymphome de Hodgkin en rechute et pour la pratique clinique, méritent d’être détaillés.
Dans les formes étendues (classification d’Ann Arbor, stades III et IV), une polychimiothérapie plus intensive par BEACOPP escaladé (bléomycine, étoposide, doxorubicine, cyclophosphamide, vincristine, procarbazine et prednisone) est recommandée chez les patients jeunes (moins de 60 ans).4 Ce traitement permet d’obtenir une rémission complète et durable chez 85 % des patients. Si une rémission complète métabolique est obtenue après deux cycles de BEACOPP escaladé, une désescalade thérapeutique vers l’ABVD est recommandée, selon les données de l’étude thérapeutique AHL 2011.4
Le traitement de la rechute repose sur une polychimiothérapie de rattrapage intensif, avec une intensification thérapeutique réalisée sous couvert d’une autogreffe de cellules souches, chez les patients éligibles à ce traitement (moins de 60 ans).2,3
En situation de rechute, des traitements ciblés (anti-CD30, brentuximab vedotin) et les immunothérapies anti-PD1 (nivolumab ou pembrolizumab) peuvent être proposés.
Les profils de tolérance du brentuximab vedotin et des anti-PD1, et la gestion des principaux effets indésirables, dans le contexte du traitement du lymphome de Hodgkin en rechute et pour la pratique clinique, méritent d’être détaillés.
Traitement par thérapie ciblée anti-CD30
La seule thérapie ciblée dans le cadre du traitement du lymphome de Hodgkin disponible et indiquée en 2023, en France, est dirigée contre l’antigène tumoral CD30 (TNF receptor superfamily member 8 [TNFRSF8]), marqueur tumoral classique des cellules du lymphome de Hodgkin. Le CD30 est la cible thérapeutique du brentuximab vedotin, un anticorps monoclonal couplé à un agent cytotoxique antimicrotubule, la monométhylauristatine E (MMAE). Le brentuximab vedotin est donc une chimiothérapie cytotoxique antimicrotubule vectorisée dirigée sur les cellules CD30 positives.5
Mécanisme d’action du brentuximab vedotin
Le brentuximab vedotin (BV) est un ADC (antibody drug conjugate, conjugué anticorps-médicament) qui libère un agent antinéoplasique, ce qui se traduit par une mort apoptotique sélective des cellules tumorales exprimant l’antigène CD30.6 Les données précliniques suggèrent que l’activité biologique du BV résulte d’un processus en plusieurs étapes. La liaison de l’ADC à l’antigène CD30 à la surface de la cellule déclenche l’internalisation du complexe ADC-CD30 qui est ensuite transféré au compartiment lysosomial.6 Au sein de la cellule, le principe actif MMAE est libéré de l’anticorps par clivage protéolytique. La liaison de la MMAE à la tubuline perturbe le réseau de microtubules dans la cellule, induit l’arrêt du cycle cellulaire et se traduit par la mort apoptotique des cellules tumorales exprimant l’antigène CD30.6
Indications et bénéfices du brentuximab vedotin
Chez les patients ayant un risque accru de récidive ou de progression après une autogreffe, ou en cas de rechute après autogreffe ou après au moins deux lignes de traitement quand la polychimiothérapie intensive n’est plus une option, un traitement ciblé sur le CD30 par BV peut être proposé.
En situation de monothérapie en troisième ligne de traitement, le taux de réponse global est de 75 %, mais la durée médiane de réponse est relativement courte (6,7 mois).4 Une minorité de patients (22 %) garde une réponse complète de qualité et maintenue après cinq ans de suivi.5
Chez les patients ayant un lymphome de Hodgkin classique réfractaire ou en rechute CD30 positif avec facteurs de risque, un traitement d’entretien par BV administré en post-autogreffe améliore significativement la survie sans progression (médiane de survie sans progression de 42,9 mois dans le bras BV versus 24,1 mois dans le bras placebo).3 Ces facteurs de risque reconnus sont un antécédent de maladie réfractaire à la chimiothérapie ou une rechute, ou une progression de la maladie dans les douze mois suivant le traitement de première ligne, ou une atteinte extraganglionnaire au moment de la rechute pré-greffe autologue de cellules souches (ASCT).7
Le BV est également approuvé dans le traitement du lymphome de Hodgkin CD30-positif de stade IV, chez les patients adultes non précédemment traités, en association avec la doxorubicine, la vinblastine et la dacarbazine (AVD) ; néanmoins, ce traitement n’est pas remboursé actuellement en France dans le cadre de la première ligne (service médical rendu insuffisant).
Le BV peut également être utilisé en première rechute en association à la chimiothérapie de rattrapage, en vue d’un projet d’autogreffe ; il s’agit d’une recommandation temporaire d’utilisation (RTU) dans l’indication en seconde ligne avant ASCT, en association à la chimiothérapie standard chez les enfants, les adolescents et les adultes.8,9 Les études thérapeutiques, principalement de phase II, montrent une augmentation du taux de rémission complète, ce qui permet d’augmenter le nombre de patients pouvant bénéficier d’une autogreffe.
La dose recommandée de BV en monothérapie est de 1,8 mg/kg, administrée par perfusion intraveineuse de trente minutes toutes les trois semaines, avec un maximum de seize cures de traitement (soit approximativement un an) si la tolérance le permet. Les patients qui obtiennent au moins une stabilisation de la maladie doivent recevoir un minimum de huit cycles et un maximum de seize cycles.5,10
En situation de monothérapie en troisième ligne de traitement, le taux de réponse global est de 75 %, mais la durée médiane de réponse est relativement courte (6,7 mois).4 Une minorité de patients (22 %) garde une réponse complète de qualité et maintenue après cinq ans de suivi.5
Chez les patients ayant un lymphome de Hodgkin classique réfractaire ou en rechute CD30 positif avec facteurs de risque, un traitement d’entretien par BV administré en post-autogreffe améliore significativement la survie sans progression (médiane de survie sans progression de 42,9 mois dans le bras BV versus 24,1 mois dans le bras placebo).3 Ces facteurs de risque reconnus sont un antécédent de maladie réfractaire à la chimiothérapie ou une rechute, ou une progression de la maladie dans les douze mois suivant le traitement de première ligne, ou une atteinte extraganglionnaire au moment de la rechute pré-greffe autologue de cellules souches (ASCT).7
Le BV est également approuvé dans le traitement du lymphome de Hodgkin CD30-positif de stade IV, chez les patients adultes non précédemment traités, en association avec la doxorubicine, la vinblastine et la dacarbazine (AVD) ; néanmoins, ce traitement n’est pas remboursé actuellement en France dans le cadre de la première ligne (service médical rendu insuffisant).
Le BV peut également être utilisé en première rechute en association à la chimiothérapie de rattrapage, en vue d’un projet d’autogreffe ; il s’agit d’une recommandation temporaire d’utilisation (RTU) dans l’indication en seconde ligne avant ASCT, en association à la chimiothérapie standard chez les enfants, les adolescents et les adultes.8,9 Les études thérapeutiques, principalement de phase II, montrent une augmentation du taux de rémission complète, ce qui permet d’augmenter le nombre de patients pouvant bénéficier d’une autogreffe.
La dose recommandée de BV en monothérapie est de 1,8 mg/kg, administrée par perfusion intraveineuse de trente minutes toutes les trois semaines, avec un maximum de seize cures de traitement (soit approximativement un an) si la tolérance le permet. Les patients qui obtiennent au moins une stabilisation de la maladie doivent recevoir un minimum de huit cycles et un maximum de seize cycles.5,10
Profil de tolérance du brentuximab vedotin
Les principaux effets indésirables du BV sont la neutropénie (de grades 3 et plus chez 13 % des patients), les infections (graves ou opportunistes chez 10 % des patients), la neuropathie périphérique (57 % des patients), les réactions à la perfusion (12 % des patients).10
Dans les essais cliniques, l’incidence d’une réaction d’hypersensibilité est de 1,2 %.11 La réaction anaphylactique survient typiquement au cours du cycle 2 de BV (au début de la perfusion) donc après une immunisation préalable et au moment de la deuxième exposition au produit comme toutes les réactions de type immuno-allergiques. En cas de réaction anaphylactique sévère, le traitement par brentuximab vedotin doit être arrêté immédiatement et un traitement médical approprié doit être administré et basé sur la prise en charge de l’anaphylaxie qui, en cas de réaction sévère, peut nécessiter de l’adrénaline et des soins intensifs. La désensibilisation au médicament pourrait être envisagé auprès d’une équipe spécialisée, par exemple en 12 ou 16 étapes et permettre une administration tolérable du brentuximab vedotin après une anaphylaxie modérée à sévère.11
En pratique clinique, l’effet indésirable le plus fréquent est la neuropathie périphérique (NP), sensitive ou sensitivomotrice (13 % des patients). Elle peut conduire à une interruption de traitement, un report de l’administration ou une diminution de dose.10
Chez les patients ayant développé une NP, le délai d’apparition médian était de douze semaines (soit au moment de la troisième cure). Chez les patients ayant présenté une NP dans les études pivots de phase II (CG035-003 et CG035-004) et dans les études de phase III en monothérapie randomisées (SGN35-005 et C25001), une résolution ou une amélioration des symptômes de la neuropathie périphérique était notée chez la plupart des patients (82 %). L’amélioration était constatée entre seize et vingt-quatre semaines après son apparition.10
Il n’y a pas, actuellement, de traitement spécifique de la NP. Sa gestion en pratique clinique est essentiellement préventive et repose sur le report de traitement, son interruption ou les baisses de doses en fonction de l’évaluation neurologique clinique. Les patients ayant une NP préexistante (post-thérapeutique liée à la chimiothérapie, ou d’une autre cause comme la neuropathie diabétique) doivent être surveillés attentivement pendant le traitement par BV.10 La gabapentine, la prégabaline et l’amitriptyline ne sont pas recommandées dans cette indication. La gestion de la NP dépend des modalités d’utilisation du BV : en monothérapie ou en combinaison avec la chimiothérapie (se référer au résumé des caractéristiques du produit pour les informations détaillées).
Letableau résume les éléments pratiques de gestion des NP liées au BV en monothérapie.
Dans les essais cliniques, l’incidence d’une réaction d’hypersensibilité est de 1,2 %.11 La réaction anaphylactique survient typiquement au cours du cycle 2 de BV (au début de la perfusion) donc après une immunisation préalable et au moment de la deuxième exposition au produit comme toutes les réactions de type immuno-allergiques. En cas de réaction anaphylactique sévère, le traitement par brentuximab vedotin doit être arrêté immédiatement et un traitement médical approprié doit être administré et basé sur la prise en charge de l’anaphylaxie qui, en cas de réaction sévère, peut nécessiter de l’adrénaline et des soins intensifs. La désensibilisation au médicament pourrait être envisagé auprès d’une équipe spécialisée, par exemple en 12 ou 16 étapes et permettre une administration tolérable du brentuximab vedotin après une anaphylaxie modérée à sévère.11
En pratique clinique, l’effet indésirable le plus fréquent est la neuropathie périphérique (NP), sensitive ou sensitivomotrice (13 % des patients). Elle peut conduire à une interruption de traitement, un report de l’administration ou une diminution de dose.10
Chez les patients ayant développé une NP, le délai d’apparition médian était de douze semaines (soit au moment de la troisième cure). Chez les patients ayant présenté une NP dans les études pivots de phase II (CG035-003 et CG035-004) et dans les études de phase III en monothérapie randomisées (SGN35-005 et C25001), une résolution ou une amélioration des symptômes de la neuropathie périphérique était notée chez la plupart des patients (82 %). L’amélioration était constatée entre seize et vingt-quatre semaines après son apparition.10
Il n’y a pas, actuellement, de traitement spécifique de la NP. Sa gestion en pratique clinique est essentiellement préventive et repose sur le report de traitement, son interruption ou les baisses de doses en fonction de l’évaluation neurologique clinique. Les patients ayant une NP préexistante (post-thérapeutique liée à la chimiothérapie, ou d’une autre cause comme la neuropathie diabétique) doivent être surveillés attentivement pendant le traitement par BV.10 La gabapentine, la prégabaline et l’amitriptyline ne sont pas recommandées dans cette indication. La gestion de la NP dépend des modalités d’utilisation du BV : en monothérapie ou en combinaison avec la chimiothérapie (se référer au résumé des caractéristiques du produit pour les informations détaillées).
Le
Immunothérapies par inhibiteurs des points de contrôle anti-PD1
Indications et bénéfices des anti-PD1
Chez les patients ayant une rechute après chimiothérapie intensive incluant une autogreffe pour ceux éligibles à ce traitement, et après échec du BV, une immunothérapie anti-PD1 peut être proposée.2,3 Les deux anti-PD1 approuvés dans le traitement du lymphome de Hodgkin sont le nivolumab et le pembrolizumab, administrés par voie intraveineuse. Les anti-PD1 sont des immunothérapies de rattrapage efficaces dans le traitement du lymphome de Hodgkin récidivant. Administrés seuls, nivolumab et pembrolizumab ont des taux de réponse élevés, mais la plupart des patients n’atteignent qu’une réponse partielle (ORR [objective response rate] : ~ 65 % ; taux de réponse complète : 10 %) chez les patients qui ont rechuté ou progressé après une ASCT.2
Fait important, les réponses thérapeutiques peuvent être prolongées, avec une durée médiane de réponse d’environ un an et surtout une phase de plateau.3 Les patients ayant obtenu une réponse complète sous traitement peuvent la maintenir et ne pas rechuter, avec des données publiées qui dépassent maintenant la médiane des cinq ans de suivi.12,13
Fait important, les réponses thérapeutiques peuvent être prolongées, avec une durée médiane de réponse d’environ un an et surtout une phase de plateau.3 Les patients ayant obtenu une réponse complète sous traitement peuvent la maintenir et ne pas rechuter, avec des données publiées qui dépassent maintenant la médiane des cinq ans de suivi.12,13
Profil de tolérance des anti-PD1
En dehors du lymphome de Hodgkin, les anti-PD1 possèdent de nombreuses indications oncologiques : mélanome, cancer bronchique, mésothéliome, carcinome rénal, cancer épidermoïde de la tête et du cou, carcinome urothélial, cancer colorectal avec instabilité microsatellitaire, carcinome épidermoïde de l’œsophage, adénocarcinome gastrique, carcinome épidermoïde cutané…14 Le recul et le suivi des patients dépassent maintenant les dix ans dans les indications oncologiques et, dans les données rapportées (encore peu nombreuses), il n’y a pas de toxicités d’organe retardées, ni de risque de second cancer, ni de développement de maladie chronique auto-immune.15,16
Dans le cadre du traitement du lymphome de Hodgkin, le profil de tolérance des anti-PD1 est globalement similaire à celui observé dans les autres indications oncologiques.12,13 La seule différence que l’on peut retrouver est l’absence d’hyperprogression induite par anti-PD1, alors que des hyperprogressions sont rapportées chez 5 à 10 % des patients en oncologie.17
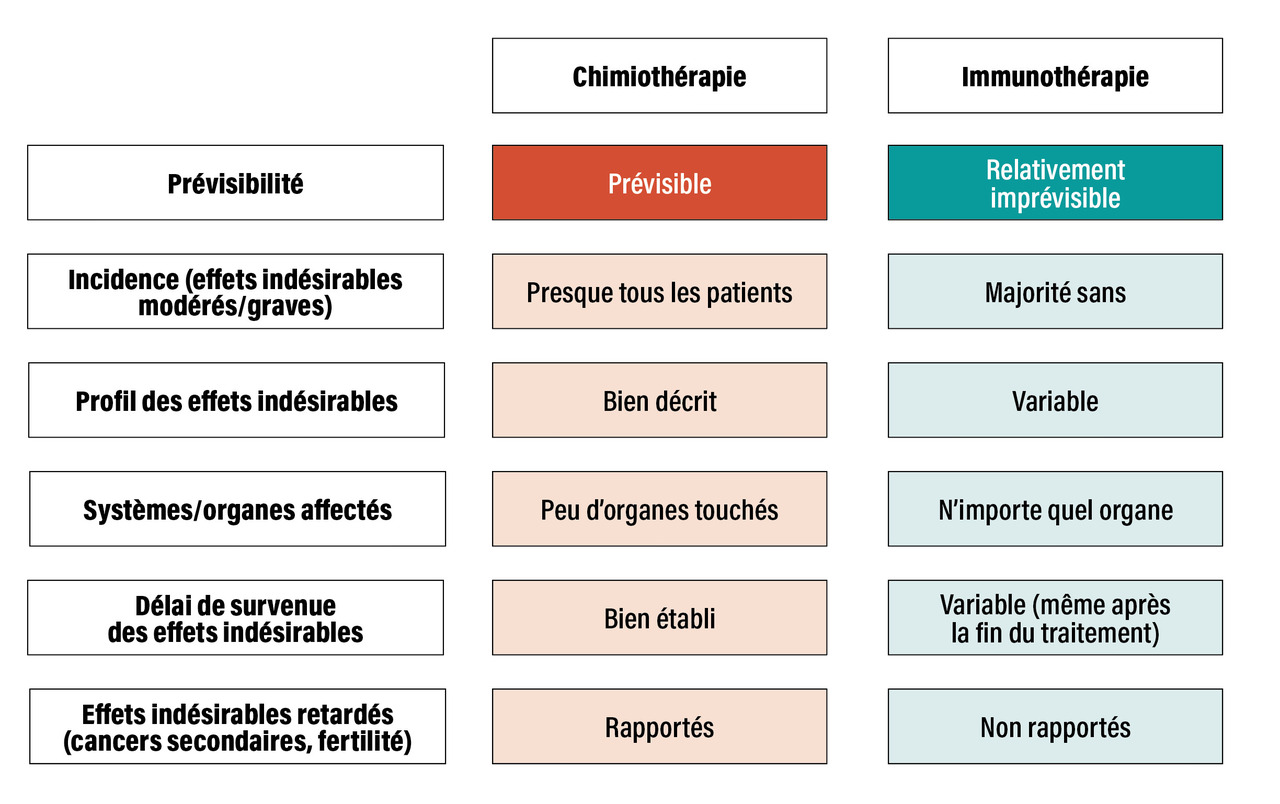
L’immunothérapie anti-PD1 est globalement mieux tolérée que la chimiothérapie, sur la base de la fréquence et de la sévérité des effets indésirables :14 les effets indésirables sévères et non sévères sont moins fréquents avec les anti-PD1 comparativement à la chimiothérapie cytotoxique ; liés à l’immunité, ils sont différents de ceux rapportés avec la chimiothérapie.14
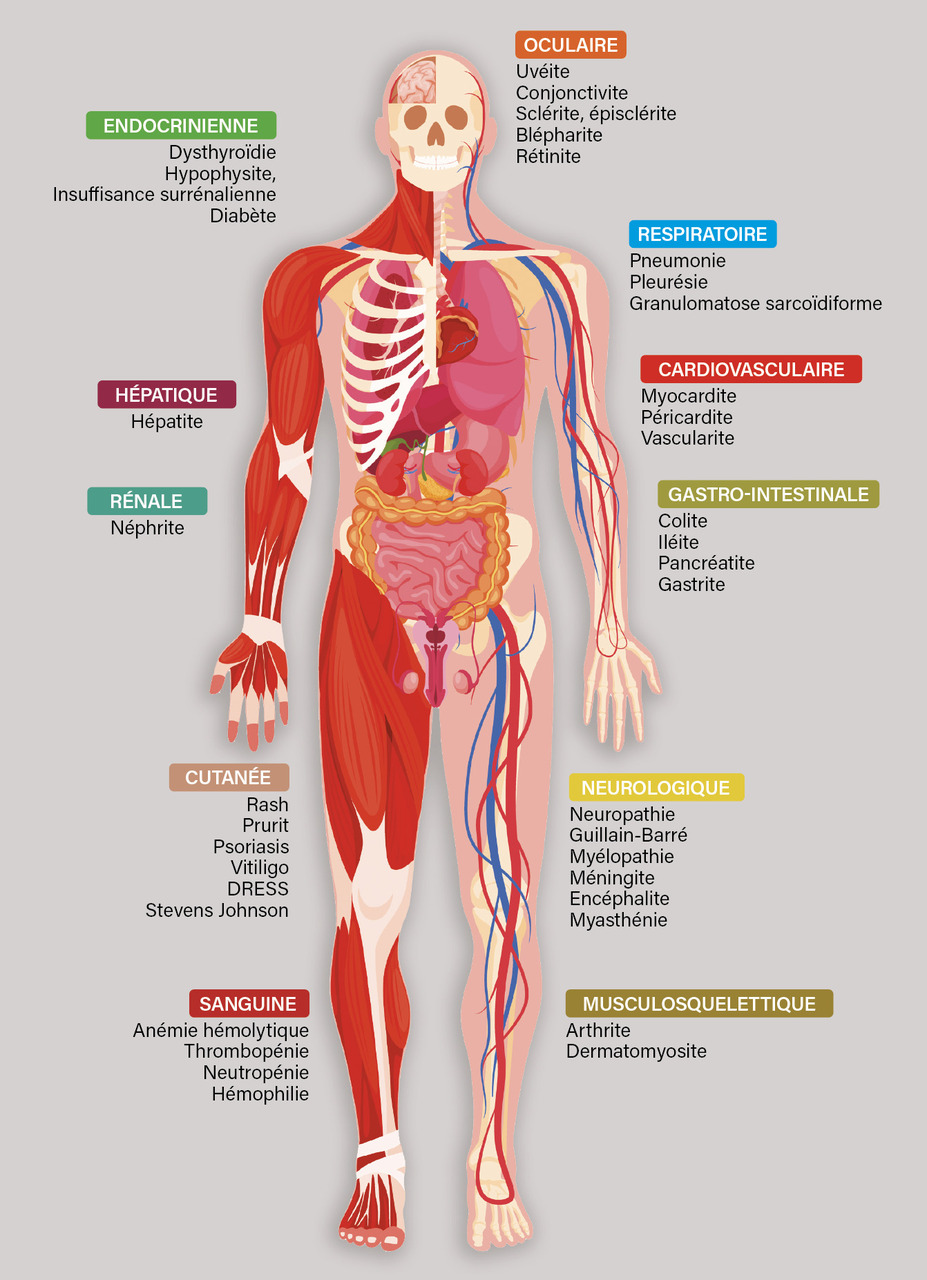
Le profil de tolérance des anti-PD1 est très varié (endocrinopathies, rash, colite, pneumopathies interstitielles), pratiquement tous les organes pouvant être concernés par une réaction immunologique dont la survenue peut être imprévisible ; les effets indésirables peuvent apparaître parfois même après l’arrêt du traitement (fig. 1 et 2).
Dans le cadre du traitement du lymphome de Hodgkin, le profil de tolérance des anti-PD1 est globalement similaire à celui observé dans les autres indications oncologiques.12,13 La seule différence que l’on peut retrouver est l’absence d’hyperprogression induite par anti-PD1, alors que des hyperprogressions sont rapportées chez 5 à 10 % des patients en oncologie.17
L’immunothérapie anti-PD1 est globalement mieux tolérée que la chimiothérapie, sur la base de la fréquence et de la sévérité des effets indésirables :14 les effets indésirables sévères et non sévères sont moins fréquents avec les anti-PD1 comparativement à la chimiothérapie cytotoxique ; liés à l’immunité, ils sont différents de ceux rapportés avec la chimiothérapie.14
Le profil de tolérance des anti-PD1 est très varié (endocrinopathies, rash, colite, pneumopathies interstitielles), pratiquement tous les organes pouvant être concernés par une réaction immunologique dont la survenue peut être imprévisible ; les effets indésirables peuvent apparaître parfois même après l’arrêt du traitement (
Situation particulière des complications d’une greffe allogénique de cellules souches hématopoïétiques
Des cas de réaction aiguë du greffon contre l’hôte (GVH) et de mortalité liée à la transplantation ont été observés durant le suivi des patients atteints de lymphome de Hodgkin ayant reçu une allogreffe après exposition antérieure au nivolumab. Une prise en compte attentive des bénéfices potentiels d’une allogreffe et d’une possible augmentation du risque de complications liées à la greffe doit être effectuée au cas par cas. Pour éviter un risque de complication immunitaire lié à la greffe, un délai minimal de « wash out » de douze semaines paraît raisonnable entre la dernière dose d’anti-PD1 et l’allogreffe.18,19
Chez les patients traités par anti-PD1 après une allogreffe, des cas sévères et rapides de réaction de greffon contre l’hôte, dont certains d’issue fatale, ont été rapportés (hors essai thérapeutique).17,18 Le traitement anti-PD1 pourrait majorer le risque de GVH sévère. Le bénéfice d’un traitement par anti-PD1 par rapport au risque de GVH doit être pris en considération chez les patients allogreffés.18,19
Chez les patients traités par anti-PD1 après une allogreffe, des cas sévères et rapides de réaction de greffon contre l’hôte, dont certains d’issue fatale, ont été rapportés (hors essai thérapeutique).17,18 Le traitement anti-PD1 pourrait majorer le risque de GVH sévère. Le bénéfice d’un traitement par anti-PD1 par rapport au risque de GVH doit être pris en considération chez les patients allogreffés.18,19
Moment de survenue et facteurs de risques des effets indésirables liés à l’immunothérapie
La médiane de survenue des effets indésirables liés à l’immunothérapie (EILI) est de dix à douze semaines après le début du traitement. Une des particularités des EILI est qu’ils peuvent survenir de façon retardée, parfois même plusieurs mois après l’arrêt du traitement. Il est donc nécessaire de maintenir une attention particulière à la survenue d’EILI même si la tolérance initiale a été bonne.14
Le principal facteur de risque de survenue d’un EILI est la présence d’un antécédent de maladie auto-immune ou inflammatoire. En cas de reprise d’une immunothérapie, les patients ayant déjà eu un EILI sont également à risque, de récidiver.20,21
De nombreux biomarqueurs sanguins prédictifs de facteurs de risque de survenue d’EILI sont à l’étude mais, à ce jour, aucun test n’est approuvé ni recommandé en pratique clinique.
Le principal facteur de risque de survenue d’un EILI est la présence d’un antécédent de maladie auto-immune ou inflammatoire. En cas de reprise d’une immunothérapie, les patients ayant déjà eu un EILI sont également à risque, de récidiver.20,21
De nombreux biomarqueurs sanguins prédictifs de facteurs de risque de survenue d’EILI sont à l’étude mais, à ce jour, aucun test n’est approuvé ni recommandé en pratique clinique.
Recommandations générales pour la gestion des EILI
En raison de la rareté des preuves de haute qualité, les recommandations de gestion des EILI sont fondées sur des consensus d’experts.22
La plupart des EILI sont peu sévères et peuvent être traités symptomatiquement. Cependant, certains peuvent directement menacer le pronostic vital et nécessitent une reconnaissance précoce pour une prise en charge spécialisée.
Bien que la prise en charge varie en fonction du système organique affecté, en général, le traitement par inhibiteur de checkpoint immunitaire (ICPi) peut être poursuivi avec une surveillance étroite des toxicités de grade 1, à l’exception des toxicités neurologiques, hématologiques et cardiaques pour lesquelles le traitement doit être interrompu en cas de toxicité de grade 1.
Le traitement par ICPi peut être suspendu pour la plupart des toxicités de grade 2, en envisageant de le reprendre lorsque les symptômes disparaissent ou reviennent au grade 1.
Le traitement de la plupart des EILI (sauf endocriniens) de grade 2 ou plus repose sur les corticostéroïdes, qui peuvent être administrés en fonction de l’organe affecté par l’EILI.22,23 Les toxicités de grade 3 justifient généralement la suspension de l’ICPi et l’initiation de corticostéroïdes à forte dose. La dose de ceux-ci doit être progressivement diminuée sur au moins quatre à six semaines.
Les corticoïdes sont efficaces pour la plupart des cas d’EILI ; certains, réfractaires à la corticothérapie, peuvent nécessiter un traitement immunosuppresseur supplémentaire et une prise en charge par un spécialiste (par exemple infliximab par le gastroentérologue). En général, l’arrêt définitif de l’ICPi est recommandé en cas de toxicités de grade 4, sauf pour les endocrinopathies contrôlées par un traitement hormonal substitutif.
La plupart des EILI sont peu sévères et peuvent être traités symptomatiquement. Cependant, certains peuvent directement menacer le pronostic vital et nécessitent une reconnaissance précoce pour une prise en charge spécialisée.
Bien que la prise en charge varie en fonction du système organique affecté, en général, le traitement par inhibiteur de checkpoint immunitaire (ICPi) peut être poursuivi avec une surveillance étroite des toxicités de grade 1, à l’exception des toxicités neurologiques, hématologiques et cardiaques pour lesquelles le traitement doit être interrompu en cas de toxicité de grade 1.
Le traitement par ICPi peut être suspendu pour la plupart des toxicités de grade 2, en envisageant de le reprendre lorsque les symptômes disparaissent ou reviennent au grade 1.
Le traitement de la plupart des EILI (sauf endocriniens) de grade 2 ou plus repose sur les corticostéroïdes, qui peuvent être administrés en fonction de l’organe affecté par l’EILI.22,23 Les toxicités de grade 3 justifient généralement la suspension de l’ICPi et l’initiation de corticostéroïdes à forte dose. La dose de ceux-ci doit être progressivement diminuée sur au moins quatre à six semaines.
Les corticoïdes sont efficaces pour la plupart des cas d’EILI ; certains, réfractaires à la corticothérapie, peuvent nécessiter un traitement immunosuppresseur supplémentaire et une prise en charge par un spécialiste (par exemple infliximab par le gastroentérologue). En général, l’arrêt définitif de l’ICPi est recommandé en cas de toxicités de grade 4, sauf pour les endocrinopathies contrôlées par un traitement hormonal substitutif.
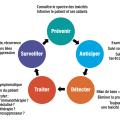
Cinq piliers temporels pour la gestion des EILI
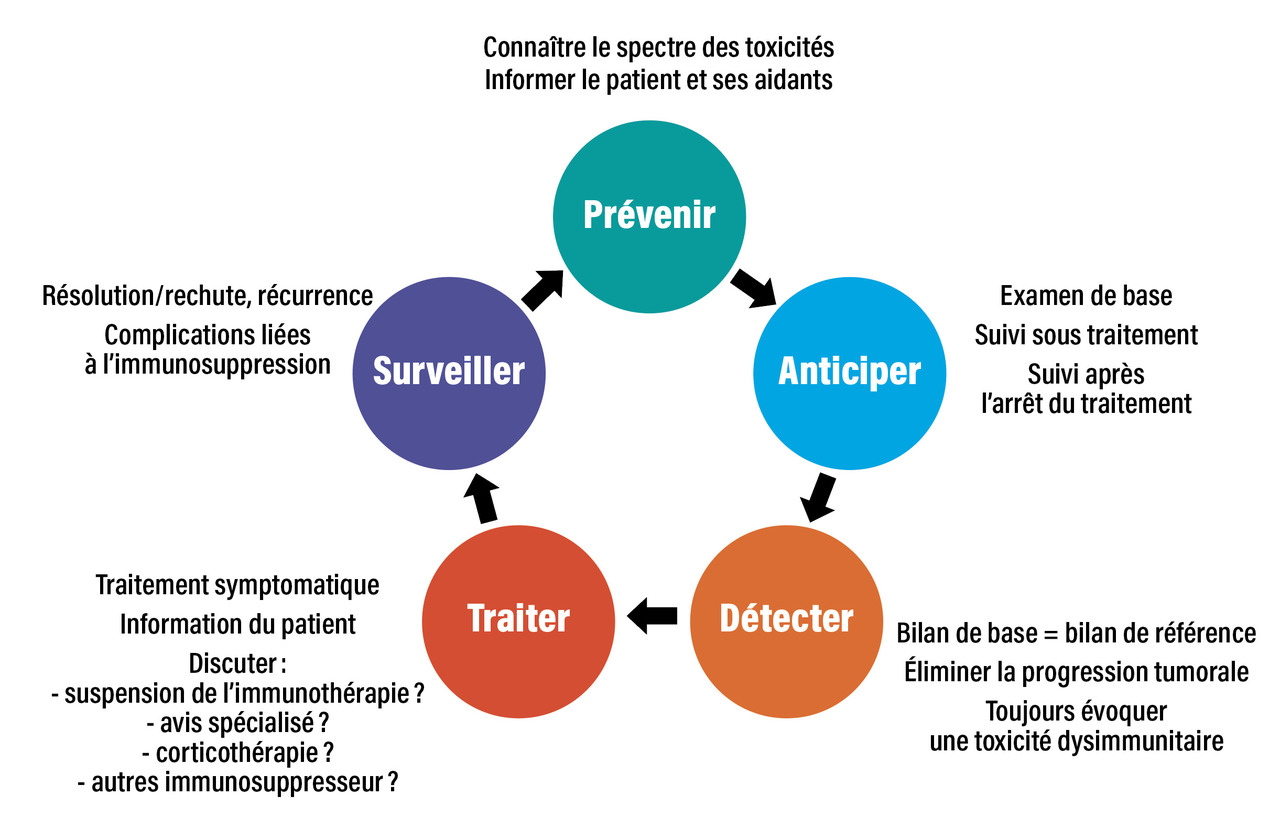
La gestion des EILI peut se résumer et se décomposer en cinq étapes successives : prévenir, anticiper, détecter, traiter, surveiller (fig. 3 ).
Prévenir
C’est probablement un des piliers les plus importants. Il s’agit d’informer le patient, son entourage mais aussi les professionnels de santé participant au parcours de soins (infirmiers, pharmaciens, médecin généraliste, urgentistes) de l’existence et de la spécificité des toxicités liées à l’immunothérapie.24 Une information claire et accessible doit être délivrée au patient, et une « carte patient sous immunothérapie » peut lui être remise pour documentation de l’information. Il convient de le prévenir que tout symptôme nouveau ou inhabituel, ou aggravation d’un symptôme préexistant doit faire craindre un effet indésirable lié à l’immunothérapie et doit être rapidement rapporté. Il est important de rappeler que la prise en charge des toxicités immunomédiées est souvent différente des toxicités habituelles liées aux traitements conventionnels (chimiothérapies, thérapies ciblées, etc.) et il convient de recommander aux patients de ne pas s’automédiquer.Anticiper
Avant l’initiation de l’immunothérapie, il est recommandé de rechercher tout antécédent personnel de pathologie auto-immune qui risquerait de s’aggraver sous traitement.24 En pratique, il convient de rechercher à l’interrogatoire des rhumatismes d’horaire inflammatoire, des diarrhées chroniques, un psoriasis, des troubles endocriniens (diabète, dysthyroïdies), ou tout autre antécédent de « maladie chronique » ou de « prise de corticoïdes/anti-inflammatoires au long cours ». Les maladies auto-immunes préexistantes ne constituent plus une contre-indication formelle pour débuter une immunothérapie mais sont une précaution d’emploi. Si elle est envisagée, l’immunothérapie doit alors s’accompagner d’une surveillance plus rapprochée et multidisciplinaire. De façon systématique avant la mise sous anti-PD1, le bilan paraclinique doit comprendre un hémogramme, un bilan hépatique (ASAT, ALAT, GGT, PAL et bilirubinémie), un ionogramme sanguin, une créatininémie, une glycémie à jeun, une bandelette urinaire ; une évaluation de la fonction thyroïdienne doit être réalisée en raison de la fréquence des dysthyroïdies, avec un dosage de la TSH et de la T4. Une imagerie pulmonaire récente (scanner en coupe fine) doit être disponible afin de servir de référence s’il survenait une toxicité pulmonaire. La réalisation des sérologies virales VIH, VHB, VHC fait partie des bonnes pratiques cliniques avant la mise sous traitement anticancéreux. Toute anomalie présente sur ce bilan préthérapeutique doit être explorée et prise en charge de façon appropriée. La recherche d’auto-anticorps systématique (comme les antinucléaires, antithyroperoxydases, antithyroglobuline…) n’est actuellement pas recommandée.Détecter
La médiane de survenue des EILI est de dix semaines, et une consultation peut être programmée pour les dépister.24 Il est souhaitable de rester attentif à la survenue d’EILI tout au long du traitement et après son arrêt, car ils peuvent survenir parfois plusieurs mois après la dernière injection. En pratique, l’apparition de nouveaux symptômes ou toute modification de l’état clinique, biologique ou radiologique par rapport au début du traitement doit faire évoquer un EILI.Traiter
La suspicion ou le diagnostic d’un EILI nécessite toujours une surveillance rapprochée afin de détecter toute aggravation ou résistance au traitement.24 Il convient d’informer le patient sur les signes de gravité devant l’amener à consulter rapidement. Lorsqu’un EILI est diagnostiqué, les différents éléments doivent être discutés, avec information du patient sur les critères de surveillance, traitement symptomatique, suspension ou arrêt de l’immunothérapie, mise en place d’une corticothérapie, recours à un avis spécialisé. En cas de difficultés diagnostiques, la suspension de l’immunothérapie doit être privilégiée afin de ne pas risquer d’aggraver un potentiel EILI.Surveiller
La plupart des EILI répondent favorablement à la suspension de l’immunothérapie et à la mise sous corticoïdes.24 Cela est particulièrement vrai pour les toxicités digestives, pulmonaires, hépatiques. Cependant, certaines toxicités peuvent s’accompagner de séquelles, comme les toxicités endocriniennes qui nécessitent la plupart du temps un traitement hormonal substitutif à vie, ou les toxicités neurologiques. Une décroissance trop rapide de la corticothérapie peut favoriser une rechute de l’EILI et nécessiter alors de remonter à une forte dose de corticoïdes afin de réaliser une décroissance encore plus progressive. La mise sous corticothérapie au long cours (≥ 1 mg/kg/j pendant au moins un mois) ou tout autre immunosuppresseur expose le patient au risque d’infection opportuniste. Dans ces situations, il convient d’instaurer une prophylaxie par triméthoprime-sulfaméthoxazole.25Prise en charge spécifique, organe par organe, des toxicités les plus fréquentes des anti-PD1
Une prise en charge détaillée des EILI est disponible dans les résumés des caractéristiques (RCP) des anti-PD1 et dans des recommandations académiques. Nous proposons ici les éléments clés de gestion des principaux EILI endocriniens, cutanés, digestifs, hépatiques, musculosquelettiques et cardiaques.
Les symptômes associés aux endocrinopathies induites par l’immunothérapie peuvent inclure des maux de tête et des changements visuels, en particulier des modifications du champ visuel liées à l’hypophysite.
Les symptômes de l’hypothyroïdie peuvent inclure une intolérance au froid, une peau sèche, une constipation, une prise de poids et/ou de la fatigue.
Des palpitations, une intolérance à la chaleur, de l’insomnie, des selles fréquentes ou une perte de poids peuvent signer une thyrotoxicose.
Des nausées, des vomissements, des douleurs abdominales, une perte de poids, des étourdissements ou une orthostasie ou une syncope, et une fatigue profonde peuvent être les symptômes d’une insuffisance surrénalienne.
Le diabète ou l’acidocétose diabétique (ACD) peut se manifester par une polyurie ou une polydipsie, des nausées ou des vomissements, des douleurs abdominales et/ou une vision floue.
La corticothérapie ne doit pas être utilisée dans le traitement des endocrinopathies liées aux anti-PD1. Les EILI endocriniens nécessitent le plus souvent une opothérapie substitutive adéquate et ne contre-indiquent pas la poursuite de l’immunothérapie, qui peut être momentanément suspendue à la phase aiguë de l’EILI. La définition des grades et des considérations supplémentaires pour la gestion des toxicités endocriniennes sont publiées dans les recommandations de l’American Society of Clinical Oncology (ASCO).22
Les éruptions cutanées ou dermatites inflammatoires englobent l’érythème polymorphe, lichénoïde, eczémateux, psoriasiforme, morbilliforme et érythrodysesthésie palmoplantaire, ou syndrome main-pied. Les symptômes associés à une éruption cutanée ou à une dermatite inflammatoire induite par l’immunothérapie peuvent varier, mais comprennent souvent des démangeaisons avec ou sans éruption cutanée, des lésions cutanées nouvelles ou aggravées, notamment des macules, des papules ou des plaques, et une perte de pigmentation cutanée. Pour les dermatoses bulleuses, les symptômes présentés peuvent également inclure des lésions cutanées nouvelles ou aggravées, y compris des bulles, une urticaire persistante ou des érosions sur la peau ou les surfaces muqueuses.
Les effets indésirables cutanés sévères comprennent le syndrome de Stevens-Johnson, la nécrolyse épidermique toxique, la pustulose exanthématique aiguë généralisée (bien que celle-ci ne soit pas toujours grave) et la réaction médicamenteuse avec éosinophilie et symptômes systémiques ou syndrome d’hypersensibilité médicamenteuse.
Les symptômes associés à la cicatrisation induite par l’immunothérapie peuvent inclure de la fièvre, une éruption cutanée généralisée, des douleurs cutanées, une desquamation de la peau, un œdème du visage ou des membres supérieurs, des pustules, des cloques ou des érosions.
Les EILI de grades 1 et 2 peuvent être pris en charge par traitement symptomatique (dermocorticoïdes, émollients et traitement antiprurigineux en privilégiant les antihistaminiques sédatifs). La classification du National Cancer Institute (NCI), le CTCAE, ne permet toutefois qu’une évaluation approximative de la toxicité dermatologique, et tout patient présentant une manifestation sévère, atypique ou persistante nécessite un avis spécialisé, au mieux avec biopsie cutanée. L’intérêt d’une corticothérapie topique (dermocorticoïdes) est à privilégier par rapport à une corticothérapie systémique. Les cas résistants aux dermocorticoïdes doivent être discutés de façon pluridisciplinaire et après un diagnostic dermatologique établi.
On considère la toxicité comme sévère (grade 3) lorsque le patient décrit une diarrhée avec au moins sept selles par jour et/ou la présence d’une douleur importante, de fièvre ou de signe d’irritation péritonéale.22,23,25
Lorsque qu’une colite induite par l’immunothérapie est suspectée, la démarche diagnostique consiste tout d’abord à éliminer une pathologie infectieuse (analyse bactériologique et virologique des selles, sans oublier l’infection à Clostridium difficile et la PCR Cytomégalovirus [CMV] dans le sang), à confirmer l’origine inflammatoire (calprotectine fécale, réalisation d’endoscopies hautes et basses avec biopsies systématiques) et à apprécier la sévérité (tomodensitométrie [TDM] abdomino-pelvienne). Les constatations endoscopiques et histologiques soulignent certaines caractéristiques communes avec les maladies inflammatoires du tube digestif, comme la rectocolite hémorragique ou la maladie de Crohn (érythème muqueux, ulcérations, infiltration lymphocytaire T et neutrophilique, abcès cryptiques, granulome…).14,27
Le traitement des formes peu sévères (grades 1 et 2) est fondé sur une corticothérapie à action intestinale (budénoside) ou une corticothérapie systémique d’intensité modérée (0,5 mg/kg de prednisone), alors que les manifestations de grades 3 et 4 ou celles de grade 2 ne répondant pas au traitement, requièrent une corticothérapie systémique à forte dose (1 mg/kg de prednisone et/ou bolus de méthylprednisolone).23 En cas de résistance à la corticothérapie après soixante-douze heures de traitement, il est recommandé de recourir à un traitement par anticorps anti-TNF alpha. L’infliximab est utilisé à la posologie de 5 à 10 mg/kg en une ou deux perfusions selon la réponse. Le védolizumab (inhibiteur de l’intégrine α4β7) pourrait être une alternative à l’infliximab, mais son délai d’action est plus long.22,28,30
Les images scanographiques les plus fréquemment rencontrées sont le verre dépoli et les condensations alvéolaires, donnant des tableaux de pneumopathie organisée, interstitielle non spécifique ou d’hypersensibilité. La démarche diagnostique consiste dans un premier temps à éliminer une origine infectieuse (Pneumocystis jirovecii, virus respiratoires, Legionella pneumophilia, Chlamydia et Mycoplasma pneumoniae), cardiaque ou une embolie pulmonaire, à réaliser une imagerie thoracique (tomodensitométrie sans et avec injection de produit de contraste), puis à effectuer, si possible, une endoscopie bronchique (avec lavage broncho-alvéolaire). La réalisation d’épreuves fonctionnelles respiratoires est également recommandée. Alors qu’une simple surveillance peut s’envisager pour les effets indésirables auto-immuns de grade 1 asymptomatiques, un recours à une corticothérapie systémique est nécessaire dans les autres cas, la dose pouvant être majorée selon la sévérité (recours à des bolus intraveineux possible). La corticorésistance est rare mais peut nécessiter un recours à d’autres immunosuppresseurs comme le cyclophosphamide.33,34 Des cas de granulome de type sarcoïdose ont également été rapportés sous ICPi ; ils sont en général peu sévères et ne nécessitent le plus souvent pas de corticothérapie.35,36
Le traitement repose, là encore, sur la corticothérapie (pouvant être associée aux immunoglobulines et/ou échanges plasmatiques). Certains patients et formes résistantes à la corticothérapie peuvent nécessiter un traitement plus intensif (bolus de cortisone, immunoglobulines par voie intraveineuse, échanges plasmatiques, cyclophosphamide…).
Toxicités endocriniennes
Les effets indésirables (EI) endocriniens liés au système immunitaire sont caractérisés par la glande ou l’organe affecté et comprennent l’hypothyroïdie primaire, la thyrotoxicose, l’insuffisance surrénalienne primaire, l’hypophysite et le diabète. Le délai médian d’apparition des toxicités endocriniennes est de 14,5 semaines (avec une fourchette large de 2 à 130 semaines).22,23,26Les symptômes associés aux endocrinopathies induites par l’immunothérapie peuvent inclure des maux de tête et des changements visuels, en particulier des modifications du champ visuel liées à l’hypophysite.
Les symptômes de l’hypothyroïdie peuvent inclure une intolérance au froid, une peau sèche, une constipation, une prise de poids et/ou de la fatigue.
Des palpitations, une intolérance à la chaleur, de l’insomnie, des selles fréquentes ou une perte de poids peuvent signer une thyrotoxicose.
Des nausées, des vomissements, des douleurs abdominales, une perte de poids, des étourdissements ou une orthostasie ou une syncope, et une fatigue profonde peuvent être les symptômes d’une insuffisance surrénalienne.
Le diabète ou l’acidocétose diabétique (ACD) peut se manifester par une polyurie ou une polydipsie, des nausées ou des vomissements, des douleurs abdominales et/ou une vision floue.
La corticothérapie ne doit pas être utilisée dans le traitement des endocrinopathies liées aux anti-PD1. Les EILI endocriniens nécessitent le plus souvent une opothérapie substitutive adéquate et ne contre-indiquent pas la poursuite de l’immunothérapie, qui peut être momentanément suspendue à la phase aiguë de l’EILI. La définition des grades et des considérations supplémentaires pour la gestion des toxicités endocriniennes sont publiées dans les recommandations de l’American Society of Clinical Oncology (ASCO).22
Toxicité cutanéo-muqueuse
Les EILI cutanés sont caractérisés comme des dermatoses inflammatoires, des dermatoses bulleuses et des effets indésirables cutanés sévères (EICS), selon le Common Terminology Criteria for Adverse Events (CTCAE). Le délai médian d’apparition des toxicités cutanées est de quatre semaines, mais il peut varier largement de deux à cent cinquante semaines.Les éruptions cutanées ou dermatites inflammatoires englobent l’érythème polymorphe, lichénoïde, eczémateux, psoriasiforme, morbilliforme et érythrodysesthésie palmoplantaire, ou syndrome main-pied. Les symptômes associés à une éruption cutanée ou à une dermatite inflammatoire induite par l’immunothérapie peuvent varier, mais comprennent souvent des démangeaisons avec ou sans éruption cutanée, des lésions cutanées nouvelles ou aggravées, notamment des macules, des papules ou des plaques, et une perte de pigmentation cutanée. Pour les dermatoses bulleuses, les symptômes présentés peuvent également inclure des lésions cutanées nouvelles ou aggravées, y compris des bulles, une urticaire persistante ou des érosions sur la peau ou les surfaces muqueuses.
Les effets indésirables cutanés sévères comprennent le syndrome de Stevens-Johnson, la nécrolyse épidermique toxique, la pustulose exanthématique aiguë généralisée (bien que celle-ci ne soit pas toujours grave) et la réaction médicamenteuse avec éosinophilie et symptômes systémiques ou syndrome d’hypersensibilité médicamenteuse.
Les symptômes associés à la cicatrisation induite par l’immunothérapie peuvent inclure de la fièvre, une éruption cutanée généralisée, des douleurs cutanées, une desquamation de la peau, un œdème du visage ou des membres supérieurs, des pustules, des cloques ou des érosions.
Les EILI de grades 1 et 2 peuvent être pris en charge par traitement symptomatique (dermocorticoïdes, émollients et traitement antiprurigineux en privilégiant les antihistaminiques sédatifs). La classification du National Cancer Institute (NCI), le CTCAE, ne permet toutefois qu’une évaluation approximative de la toxicité dermatologique, et tout patient présentant une manifestation sévère, atypique ou persistante nécessite un avis spécialisé, au mieux avec biopsie cutanée. L’intérêt d’une corticothérapie topique (dermocorticoïdes) est à privilégier par rapport à une corticothérapie systémique. Les cas résistants aux dermocorticoïdes doivent être discutés de façon pluridisciplinaire et après un diagnostic dermatologique établi.
Toxicité digestive
Les EILI gastro-intestinaux comprennent la colite, la gastrite et l’entérocolite. Le délai médian d’apparition des toxicités gastro-intestinales est de six semaines, avec un large intervalle de 1 à 107,5 semaines. Les symptômes associés à la colite peuvent inclure des douleurs abdominales, des nausées, de la diarrhée, du sang et du mucus dans les selles, et de la fièvre.On considère la toxicité comme sévère (grade 3) lorsque le patient décrit une diarrhée avec au moins sept selles par jour et/ou la présence d’une douleur importante, de fièvre ou de signe d’irritation péritonéale.22,23,25
Lorsque qu’une colite induite par l’immunothérapie est suspectée, la démarche diagnostique consiste tout d’abord à éliminer une pathologie infectieuse (analyse bactériologique et virologique des selles, sans oublier l’infection à Clostridium difficile et la PCR Cytomégalovirus [CMV] dans le sang), à confirmer l’origine inflammatoire (calprotectine fécale, réalisation d’endoscopies hautes et basses avec biopsies systématiques) et à apprécier la sévérité (tomodensitométrie [TDM] abdomino-pelvienne). Les constatations endoscopiques et histologiques soulignent certaines caractéristiques communes avec les maladies inflammatoires du tube digestif, comme la rectocolite hémorragique ou la maladie de Crohn (érythème muqueux, ulcérations, infiltration lymphocytaire T et neutrophilique, abcès cryptiques, granulome…).14,27
Le traitement des formes peu sévères (grades 1 et 2) est fondé sur une corticothérapie à action intestinale (budénoside) ou une corticothérapie systémique d’intensité modérée (0,5 mg/kg de prednisone), alors que les manifestations de grades 3 et 4 ou celles de grade 2 ne répondant pas au traitement, requièrent une corticothérapie systémique à forte dose (1 mg/kg de prednisone et/ou bolus de méthylprednisolone).23 En cas de résistance à la corticothérapie après soixante-douze heures de traitement, il est recommandé de recourir à un traitement par anticorps anti-TNF alpha. L’infliximab est utilisé à la posologie de 5 à 10 mg/kg en une ou deux perfusions selon la réponse. Le védolizumab (inhibiteur de l’intégrine α4β7) pourrait être une alternative à l’infliximab, mais son délai d’action est plus long.22,28,30
Toxicité hépatique
La survenue d’un effet indésirable auto-immun hépatique doit être suspectée devant l’apparition d’une hépatite cytolytique et/ou cholestatique isolée.31,32 Cette toxicité est le plus souvent asymptomatique, mais des cas d’hépatite fulminante ont été décrits. Le délai de survenue habituel est de six à douze semaines après l’initiation du traitement, mais des cas d’hépatites tardives (après plus d’un an) sont observés. On considère une hépatite comme sévère (grade 3) si le taux d’ASAT/ALAT est supérieur à cinq fois la normale et/ou si le taux de bilirubine totale est supérieur à trois fois la normale. Le bilan biologique et morphologique permet d’éliminer les hépatites virales, la présence de métastases, une lithiase ou une thrombose des veines sus-hépatiques. Il faut également s’assurer de l’absence d’une autre cause médicamenteuse. La biopsie hépatique doit être proposée au cas par cas, en particulier en cas de doute étiologique. L’histologie met en évidence un infiltrat lymphocytaire T panlobulaire, un infiltrat histiocytaire sinusoïdal, une atteinte des voies biliaires et des lésions des veines centrolobulaires. Une hépatite granulomateuse a également été rapportée. Des cas plus rares de cholangite sclérosante avec lésions biliaires en cholangio-IRM ont également été décrits. La prise en charge thérapeutique est sensiblement la même que celle de l’hépatite auto-immune, reposant sur la corticothérapie (orale ou intraveineuse) puis, en cas d’inefficacité après soixante-douze heures ou de corticodépendance, sur l’ajout de mycophénolate mofétil (MMF).Toxicité musculosquelettique
Les EILI musculosquelettiques sont caractérisés par l’arthrite inflammatoire, la myosite et le syndrome de type polymyalgie. Le délai médian d’apparition est de trente-huit semaines, mais peut varier considérablement de 1 à 127 semaines.22,23,26 Les symptômes peuvent inclure des douleurs articulaires accompagnées d’un gonflement articulaire et/ou de symptômes inflammatoires tels qu’une raideur après l’inactivité ou le matin, durant plus de trente minutes à une heure. Les symptômes de la myosite induite par l’immunothérapie peuvent comprendre des douleurs et une faiblesse musculaires. Les patients atteints de myosite peuvent également développer un syndrome de type myasthénie grave et/ou une myocardite, qui peuvent mettre en jeu le pronostic vital si les muscles respiratoires ou le myocarde sont impliqués. L’ASCO a publié l’ensemble des recommandations, la définition des grades et des considérations supplémentaires pour les toxicités musculosquelettiques.21 La prise en charge thérapeutique des EILI avec arthrite repose sur la corticothérapie, avec la nécessité de recourir à un agent immunosuppresseur, en particulier le méthotrexate, utile à visée d’épargne cortisonique. L’ICPi est le plus souvent poursuivi après une suspension transitoire.Toxicité pulmonaire
La pneumonie interstitielle, EILI, est définie comme une inflammation focale ou diffuse du parenchyme pulmonaire, généralement identifiée sur l’imagerie par tomodensitométrie. Il n’y a pas de caractéristiques symptomatiques, pathologiques ou radiographiques pathognomoniques ; les symptômes associés à la pneumonite induite par l’immunothérapie peuvent inclure une toux nouvelle ou aggravée, un essoufflement, un besoin accru en oxygène, des douleurs thoraciques et/ou de la fièvre.22,23,26Les images scanographiques les plus fréquemment rencontrées sont le verre dépoli et les condensations alvéolaires, donnant des tableaux de pneumopathie organisée, interstitielle non spécifique ou d’hypersensibilité. La démarche diagnostique consiste dans un premier temps à éliminer une origine infectieuse (Pneumocystis jirovecii, virus respiratoires, Legionella pneumophilia, Chlamydia et Mycoplasma pneumoniae), cardiaque ou une embolie pulmonaire, à réaliser une imagerie thoracique (tomodensitométrie sans et avec injection de produit de contraste), puis à effectuer, si possible, une endoscopie bronchique (avec lavage broncho-alvéolaire). La réalisation d’épreuves fonctionnelles respiratoires est également recommandée. Alors qu’une simple surveillance peut s’envisager pour les effets indésirables auto-immuns de grade 1 asymptomatiques, un recours à une corticothérapie systémique est nécessaire dans les autres cas, la dose pouvant être majorée selon la sévérité (recours à des bolus intraveineux possible). La corticorésistance est rare mais peut nécessiter un recours à d’autres immunosuppresseurs comme le cyclophosphamide.33,34 Des cas de granulome de type sarcoïdose ont également été rapportés sous ICPi ; ils sont en général peu sévères et ne nécessitent le plus souvent pas de corticothérapie.35,36
Toxicité neurologique
Les EILI neurologiques englobent un large éventail de syndromes, notamment la myasthénie grave ou syndrome myasthénique, la myasthénie grave avec chevauchement de myosite, la méningite aseptique, l’encéphalite, le syndrome de type Guillain-Barré et une variété d’autres phénotypes de neuropathie périphérique et des troubles démyélinisants.22,23 Les symptômes de la myasthénie grave peuvent inclure une faiblesse musculaire fatigable ou fluctuante, un ptosis, une vision double, une dysphagie, une dysarthrie, une faiblesse des muscles faciaux et/ou une chute de la tête ou une faiblesse du cou. Le syndrome de Guillain-Barré peut se manifester par une faiblesse musculaire ascendante et progressive, un essoufflement, une faiblesse faciale, un engourdissement et des picotements dans les pieds ou les mains, des sensations de brûlure, de coups de poignard ou des douleurs lancinantes dans les zones atteintes, une perte d’équilibre et de coordination.37 La méningite aseptique peut se présenter par des maux de tête, une photophobie, une raideur de la nuque, des nausées ou des vomissements et parfois de la fièvre. L’état mental doit être normal, contrairement à l’encéphalite. Les symptômes de l’encéphalite peuvent inclure une confusion, une altération de l’état mental, une altération du comportement, des maux de tête, des convulsions, une faiblesse et une instabilité de la démarche. D’autres maladies démyélinisantes potentiellement liées au système immunitaire comprennent la sclérose en plaques, la myélite transverse, l’encéphalomyélite aiguë disséminée, la névrite optique et la neuromyélite optique.37Le traitement repose, là encore, sur la corticothérapie (pouvant être associée aux immunoglobulines et/ou échanges plasmatiques). Certains patients et formes résistantes à la corticothérapie peuvent nécessiter un traitement plus intensif (bolus de cortisone, immunoglobulines par voie intraveineuse, échanges plasmatiques, cyclophosphamide…).
Toxicité cardiaque
La myocardite est le principal effet indésirable auto-immun cardiaque décrit sous ICPi. Son incidence est rare (inférieure à 1 %, survenant surtout sous bithérapie), mais elle peut être fatale et doit donc être dépistée et prise en charge précocement.38 Le délai de survenue est précoce (moins d’un mois après l’initiation du traitement). Le pronostic est sombre, puisqu’il est rapporté un taux de décès de 50 %. Les manifestations sont variables, certains patients étant asymptomatiques, d’autres présentant des signes d’insuffisance cardiaque et/ou d’arythmie (auriculaire ou ventriculaire). Les marqueurs de nécrose myocardique (troponine, CK-MB) sont constamment élevés, et l’imagerie par résonance magnétique (IRM) cardiaque permet de confirmer le diagnostic grâce à la visualisation de l’inflammation myocardique, offrant ainsi la possibilité d’éviter la plupart du temps le recours à la biopsie myocardique. Celle-ci doit rester exceptionnelle, l’histologie montrant un infiltrat lymphocytaire touchant le myocarde, le système de conduction et les fibres musculaires squelettiques. Par ailleurs, parmi les patients avec myocardite, 25 % ont une myosite associée et 10 % une myasthénie.38 La prise en charge thérapeutique consiste en un arrêt systématique de l’ICPi, et ce quel que soit le grade de la myocardite. La présence de symptômes, de signes à l’électrocardiogramme ou à l’échocardiographie, ou la présence d’une sémiologie aiguë d’inflammation à l’IRM doivent faire considérer le recours à une corticothérapie systémique.22,38 La mauvaise évolution ou l’instabilité clinique fait discuter le recours à un autre immunosuppresseur en plus des mesures de réanimation habituelles.Tolérance variable et souvent imprévisible
Les effets indésirables des thérapies ciblant le CD30 par BV sont principalement la neutropénie et la NP. La survenue d’une neuropathie chez un patient traité par BV impose une surveillance attentive et peut nécessiter une adaptation du plan de traitement. La plupart des patients récupèrent.
La tolérance des anti-PD1 est meilleure que celle de la chimiothérapie. Le profil des effets indésirables immuns-médiés reste imprévisible et varié : de nombreux organes peuvent être concernés, et leur survenue est possible même après l’arrêt de l’immunothérapie. Avec un recul de plus de dix ans, il n’y a pas de signal de toxicité retardée rapportée des anti-PD1.
La tolérance des anti-PD1 est meilleure que celle de la chimiothérapie. Le profil des effets indésirables immuns-médiés reste imprévisible et varié : de nombreux organes peuvent être concernés, et leur survenue est possible même après l’arrêt de l’immunothérapie. Avec un recul de plus de dix ans, il n’y a pas de signal de toxicité retardée rapportée des anti-PD1.
Références
1. Andre MPE, Girinsky T, Federico M, Reman O, Fortpied C, Gotti M, et al. Early positron emission tomography response-adapted treatment in stage I and II Hodgkin lymphoma: Final results of the randomized EORTC/LYSA/FIL H10 trial. J Clin Oncol 2017;35(16):1786-94.
2. Michot JM, Lazarovici J, Ghez D, Danu A, Ferme C, Bigorgne A, et al. Challenges and perspectives in the immunotherapy of Hodgkin lymphoma. Eur J Cancer. 2017;85:67-77.
3. Ansell SM. Hodgkin lymphoma: A 2020 update on diagnosis, risk-stratification, and management. Am J Hematol 2020;95(8):978-89.
4. Casasnovas RO, Bouabdallah R, Brice P, Lazarovici J, Ghesquieres H, Stamatoullas A, et al. PET-adapted treatment for newly diagnosed advanced Hodgkin lymphoma (AHL2011): A randomised, multicentre, non-inferiority, phase 3 study. Lancet Oncol 2019;20(2):202-15.
5. Moskowitz AJ. Optimizing the role of brentuximab vedotin in classical Hodgkin lymphoma therapy. Hematology Am Soc Hematol Educ Program 2018;2018(1):207-12.
6. Choi Y, Diefenbach CS. An evaluation of brentuximab vedotin as a treatment option for stage III/IV Hodgkin lymphoma. Expert Rev Hematol 2019;12(10):801-8.
7. Eichenauer DA, Aleman BMP, Andre M, Federico M, Hutchings M, Illidge T, et al. Hodgkin lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018;29(Suppl 4):iv19-iv29.
8. Stamatoullas A, Ghesquieres H, Feugier P, Andre M, Le Bras F, Gac AC, et al. Final results of brentuximab vedotin combined with ifosfamide-carboplatin-etoposide in first refractory/relapsed Hodgkin lymphoma: A lymphoma study association phase I/II study. Leuk Lymphoma 2022;63(13):3063-71.
9. Kersten MJ, Driessen J, Zijlstra JM, Plattel WJ, Morschhauser F, Lugtenburg PJ, et al. Combining brentuximab vedotin with dexamethasone, high-dose cytarabine and cisplatin as salvage treatment in relapsed or refractory Hodgkin lymphoma: The phase II HOVON/LLPC Transplant BRaVE study. Haematologica 2021;106(4):1129-37.
10. Scott LJ. Brentuximab vedotin: A review in CD30-positive Hodgkin lymphoma. Drugs 2017;77(4):435-45.
11. Villarreal-González RV, González-Díaz SN, Santos-Fernández WJ, Colunga-Pedraza PR, Varela-Constantino AL, Gómez-Almaguer D. Desensitization to brentuximab vedotin after anaphylaxis in refractory Hodgkin’s lymphoma. J Oncol Pharm Pract 2022;10781552221074965.
12. Armand P, Kuruvilla J, Michot JM, Ribrag V, Zinzani PL, Zhu Y, et al. KEYNOTE-013 4-year follow-up of pembrolizumab in classical Hodgkin lymphoma after brentuximab vedotin failure. Blood Adv 2020;4(12):2617-22.
13. Armand P, Engert A, Younes A, Fanale M, Santoro A, Zinzani PL, et al. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: Extended follow-up of the multicohort single-arm phase II checkmate 205 trial. J Clin Oncol 2018;36(14):1428-39.
14. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur J Cancer 2016;54:139-48.
15. Gauci ML, Lanoy E, Champiat S, Caramella C, Ammari S, Aspeslagh S, et al. Long-term survival in patients responding to anti-PD-1/PD-L1 therapy and disease outcome upon treatment discontinuation. Clin Cancer Res 2019;25(3): 946-56.
16. Domnariu PA, Noel N, Hardy-Leger I, Michot JM, Lambotte O. Long-term impact of immunotherapy on quality of life of surviving patients: A multi-dimensional descriptive clinical study. Eur J Cancer 2021;148:211-4.
17. Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria JC, et al. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat Rev Clin Oncol 2018;15(12):748-62.
18. Dada R, Usman B. Allogeneic hematopoietic stem cell transplantation in r/r Hodgkin lymphoma after treatment with checkpoint inhibitors: Feasibility and safety. Eur J Haematol 2019;102(2):150-6.
19. Ijaz A, Khan AY, Malik SU, Faridi W, Fraz MA, Usman M, et al. Significant risk of graft-versus-host disease with exposure to checkpoint inhibitors before and after allogeneic transplantation. Biol Blood Marrow Transplant 2019;25(1): 94-9.
20. Menzies AM, Johnson DB, Ramanujam S, Atkinson VG, Wong ANM, Park JJ, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol 2017;28(2):368-76.
21. Michot JM, Lappara A, Le Pavec J, Simonaggio A, Collins M, De Martin E, et al. The 2016-2019 ImmunoTOX assessment board report of collaborative management of immune-related adverse events, an observational clinical study. Eur J Cancer 2020;130:39-50.
22. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2018;36(17):1714-68.
23. Brahmer JR, Abu-Sbeih H, Ascierto PA, Brufsky J, Cappelli LC, Cortazar FB, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J Immunother Cancer 2021;9(6).
24. Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F, et al. Management of immune checkpoint blockade dysimmune toxicities: A collaborative position paper. Ann Oncol 2016;27(4):559-74.
25. Aldea M, Orillard E, Mansi L, Marabelle A, Scotte F, Lambotte O, et al. How to manage patients with corticosteroids in oncology in the era of immunotherapy? Eur J Cancer 2020;141:239-51.
26. Thompson JA, Schneider BJ, Brahmer J, Andrews S, Armand P, Bhatia S, et al. Management of immunotherapy-related toxicities, version 1.2019. J Natl Compr Canc Netw 2019;17(3):255-89.
27. Haanen J, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2017;28(suppl4):iv119-iv42.
28. Luo J, Beattie JA, Fuentes P, Rizvi H, Egger JV, Kern JA, et al. Beyond steroids: Immunosuppressants in steroid-refractory or resistant immune-related adverse events. J Thorac Oncol 2021;16(10):1759-64.
29. Beaugerie L, Kirchgesner J. Balancing benefit vs risk of immunosuppressive therapy for individual patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol 2019;17(3):370-9.
30. Esfahani K, Elkrief A, Calabrese C, Lapointe R, Hudson M, Routy B, et al. Moving towards personalized treatments of immune-related adverse events. Nat Rev Clin Oncol 2020;17(8):504-15.
31. De Martin E, Michot JM, Rosmorduc O, Guettier C, Samuel D. Liver toxicity as a limiting factor to the increasing use of immune checkpoint inhibitors. JHEP Rep 2020;2(6):100170.
32. Cheung V, Gupta T, Payne M, Middleton MR, Collier JD, Simmons A, et al. Immunotherapy-related hepatitis: Real-world experience from a tertiary centre. Frontline Gastroenterol. 2019;10(4):364-71.
33. Camard M, Besse B, Cariou PL, Massayke S, Laparra A, Noel N, et al. Prevalence and outcome of steroid-resistant/refractory pneumonitis induced by immune checkpoint inhibitors. Respir Med Res 2022;82:100969.
34. Balaji A, Hsu M, Lin CT, Feliciano J, Marrone K, Brahmer JR, et al. Steroid-refractory PD-(L)1 pneumonitis: Incidence, clinical features, treatment, and outcomes. J Immunother Cancer 2021;9(1).
35. Cabanie C, Ammari S, Hans S, Pobel C, Laparra A, Danlos FX, et al. Outcomes of patients with cancer and sarcoid-like granulomatosis associated with immune checkpoint inhibitors: A case-control study. Eur J Cancer 2021;156:46-59.
36. Chanson N, Ramos-Casals M, Pundole X, Suijkerbuijk K, Jose de Barros ESM, Lidar M, et al. Immune checkpoint inhibitor-associated sarcoidosis: A usually benign disease that does not require immunotherapy discontinuation. Eur J Cancer 2021;158:208-16.
37. Placais L, Michot JM, Champiat S, Romano-Martin P, Baldini C, Joao MS, et al. Neurological complications induced by immune checkpoint inhibitors: A comprehensive descriptive case-series unravelling high risk of long-term sequelae. Brain Commun2021;3(4):fcab220.
38. Moslehi J, Salem JE. Immune checkpoint inhibitor myocarditis treatment strategies and future directions. JACC CardioOncol 2022;4(5):704-7.
2. Michot JM, Lazarovici J, Ghez D, Danu A, Ferme C, Bigorgne A, et al. Challenges and perspectives in the immunotherapy of Hodgkin lymphoma. Eur J Cancer. 2017;85:67-77.
3. Ansell SM. Hodgkin lymphoma: A 2020 update on diagnosis, risk-stratification, and management. Am J Hematol 2020;95(8):978-89.
4. Casasnovas RO, Bouabdallah R, Brice P, Lazarovici J, Ghesquieres H, Stamatoullas A, et al. PET-adapted treatment for newly diagnosed advanced Hodgkin lymphoma (AHL2011): A randomised, multicentre, non-inferiority, phase 3 study. Lancet Oncol 2019;20(2):202-15.
5. Moskowitz AJ. Optimizing the role of brentuximab vedotin in classical Hodgkin lymphoma therapy. Hematology Am Soc Hematol Educ Program 2018;2018(1):207-12.
6. Choi Y, Diefenbach CS. An evaluation of brentuximab vedotin as a treatment option for stage III/IV Hodgkin lymphoma. Expert Rev Hematol 2019;12(10):801-8.
7. Eichenauer DA, Aleman BMP, Andre M, Federico M, Hutchings M, Illidge T, et al. Hodgkin lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018;29(Suppl 4):iv19-iv29.
8. Stamatoullas A, Ghesquieres H, Feugier P, Andre M, Le Bras F, Gac AC, et al. Final results of brentuximab vedotin combined with ifosfamide-carboplatin-etoposide in first refractory/relapsed Hodgkin lymphoma: A lymphoma study association phase I/II study. Leuk Lymphoma 2022;63(13):3063-71.
9. Kersten MJ, Driessen J, Zijlstra JM, Plattel WJ, Morschhauser F, Lugtenburg PJ, et al. Combining brentuximab vedotin with dexamethasone, high-dose cytarabine and cisplatin as salvage treatment in relapsed or refractory Hodgkin lymphoma: The phase II HOVON/LLPC Transplant BRaVE study. Haematologica 2021;106(4):1129-37.
10. Scott LJ. Brentuximab vedotin: A review in CD30-positive Hodgkin lymphoma. Drugs 2017;77(4):435-45.
11. Villarreal-González RV, González-Díaz SN, Santos-Fernández WJ, Colunga-Pedraza PR, Varela-Constantino AL, Gómez-Almaguer D. Desensitization to brentuximab vedotin after anaphylaxis in refractory Hodgkin’s lymphoma. J Oncol Pharm Pract 2022;10781552221074965.
12. Armand P, Kuruvilla J, Michot JM, Ribrag V, Zinzani PL, Zhu Y, et al. KEYNOTE-013 4-year follow-up of pembrolizumab in classical Hodgkin lymphoma after brentuximab vedotin failure. Blood Adv 2020;4(12):2617-22.
13. Armand P, Engert A, Younes A, Fanale M, Santoro A, Zinzani PL, et al. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: Extended follow-up of the multicohort single-arm phase II checkmate 205 trial. J Clin Oncol 2018;36(14):1428-39.
14. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur J Cancer 2016;54:139-48.
15. Gauci ML, Lanoy E, Champiat S, Caramella C, Ammari S, Aspeslagh S, et al. Long-term survival in patients responding to anti-PD-1/PD-L1 therapy and disease outcome upon treatment discontinuation. Clin Cancer Res 2019;25(3): 946-56.
16. Domnariu PA, Noel N, Hardy-Leger I, Michot JM, Lambotte O. Long-term impact of immunotherapy on quality of life of surviving patients: A multi-dimensional descriptive clinical study. Eur J Cancer 2021;148:211-4.
17. Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria JC, et al. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat Rev Clin Oncol 2018;15(12):748-62.
18. Dada R, Usman B. Allogeneic hematopoietic stem cell transplantation in r/r Hodgkin lymphoma after treatment with checkpoint inhibitors: Feasibility and safety. Eur J Haematol 2019;102(2):150-6.
19. Ijaz A, Khan AY, Malik SU, Faridi W, Fraz MA, Usman M, et al. Significant risk of graft-versus-host disease with exposure to checkpoint inhibitors before and after allogeneic transplantation. Biol Blood Marrow Transplant 2019;25(1): 94-9.
20. Menzies AM, Johnson DB, Ramanujam S, Atkinson VG, Wong ANM, Park JJ, et al. Anti-PD-1 therapy in patients with advanced melanoma and preexisting autoimmune disorders or major toxicity with ipilimumab. Ann Oncol 2017;28(2):368-76.
21. Michot JM, Lappara A, Le Pavec J, Simonaggio A, Collins M, De Martin E, et al. The 2016-2019 ImmunoTOX assessment board report of collaborative management of immune-related adverse events, an observational clinical study. Eur J Cancer 2020;130:39-50.
22. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2018;36(17):1714-68.
23. Brahmer JR, Abu-Sbeih H, Ascierto PA, Brufsky J, Cappelli LC, Cortazar FB, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J Immunother Cancer 2021;9(6).
24. Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F, et al. Management of immune checkpoint blockade dysimmune toxicities: A collaborative position paper. Ann Oncol 2016;27(4):559-74.
25. Aldea M, Orillard E, Mansi L, Marabelle A, Scotte F, Lambotte O, et al. How to manage patients with corticosteroids in oncology in the era of immunotherapy? Eur J Cancer 2020;141:239-51.
26. Thompson JA, Schneider BJ, Brahmer J, Andrews S, Armand P, Bhatia S, et al. Management of immunotherapy-related toxicities, version 1.2019. J Natl Compr Canc Netw 2019;17(3):255-89.
27. Haanen J, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2017;28(suppl4):iv119-iv42.
28. Luo J, Beattie JA, Fuentes P, Rizvi H, Egger JV, Kern JA, et al. Beyond steroids: Immunosuppressants in steroid-refractory or resistant immune-related adverse events. J Thorac Oncol 2021;16(10):1759-64.
29. Beaugerie L, Kirchgesner J. Balancing benefit vs risk of immunosuppressive therapy for individual patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol 2019;17(3):370-9.
30. Esfahani K, Elkrief A, Calabrese C, Lapointe R, Hudson M, Routy B, et al. Moving towards personalized treatments of immune-related adverse events. Nat Rev Clin Oncol 2020;17(8):504-15.
31. De Martin E, Michot JM, Rosmorduc O, Guettier C, Samuel D. Liver toxicity as a limiting factor to the increasing use of immune checkpoint inhibitors. JHEP Rep 2020;2(6):100170.
32. Cheung V, Gupta T, Payne M, Middleton MR, Collier JD, Simmons A, et al. Immunotherapy-related hepatitis: Real-world experience from a tertiary centre. Frontline Gastroenterol. 2019;10(4):364-71.
33. Camard M, Besse B, Cariou PL, Massayke S, Laparra A, Noel N, et al. Prevalence and outcome of steroid-resistant/refractory pneumonitis induced by immune checkpoint inhibitors. Respir Med Res 2022;82:100969.
34. Balaji A, Hsu M, Lin CT, Feliciano J, Marrone K, Brahmer JR, et al. Steroid-refractory PD-(L)1 pneumonitis: Incidence, clinical features, treatment, and outcomes. J Immunother Cancer 2021;9(1).
35. Cabanie C, Ammari S, Hans S, Pobel C, Laparra A, Danlos FX, et al. Outcomes of patients with cancer and sarcoid-like granulomatosis associated with immune checkpoint inhibitors: A case-control study. Eur J Cancer 2021;156:46-59.
36. Chanson N, Ramos-Casals M, Pundole X, Suijkerbuijk K, Jose de Barros ESM, Lidar M, et al. Immune checkpoint inhibitor-associated sarcoidosis: A usually benign disease that does not require immunotherapy discontinuation. Eur J Cancer 2021;158:208-16.
37. Placais L, Michot JM, Champiat S, Romano-Martin P, Baldini C, Joao MS, et al. Neurological complications induced by immune checkpoint inhibitors: A comprehensive descriptive case-series unravelling high risk of long-term sequelae. Brain Commun2021;3(4):fcab220.
38. Moslehi J, Salem JE. Immune checkpoint inhibitor myocarditis treatment strategies and future directions. JACC CardioOncol 2022;4(5):704-7.