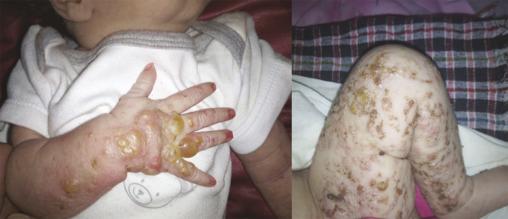
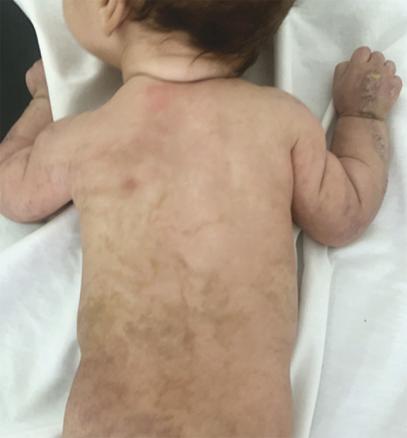
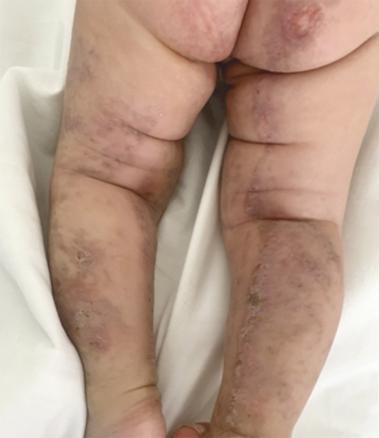
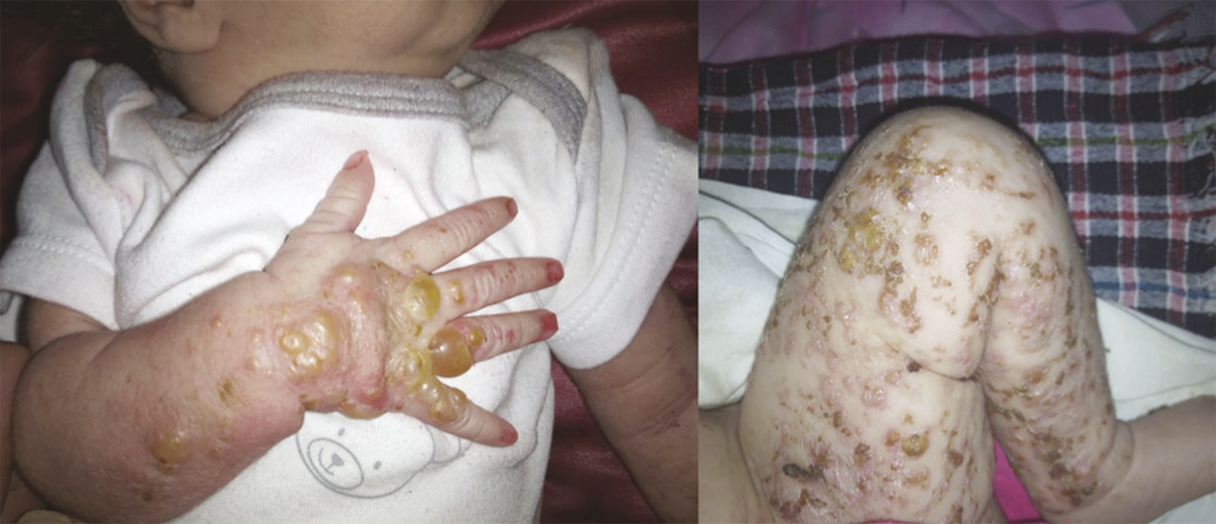
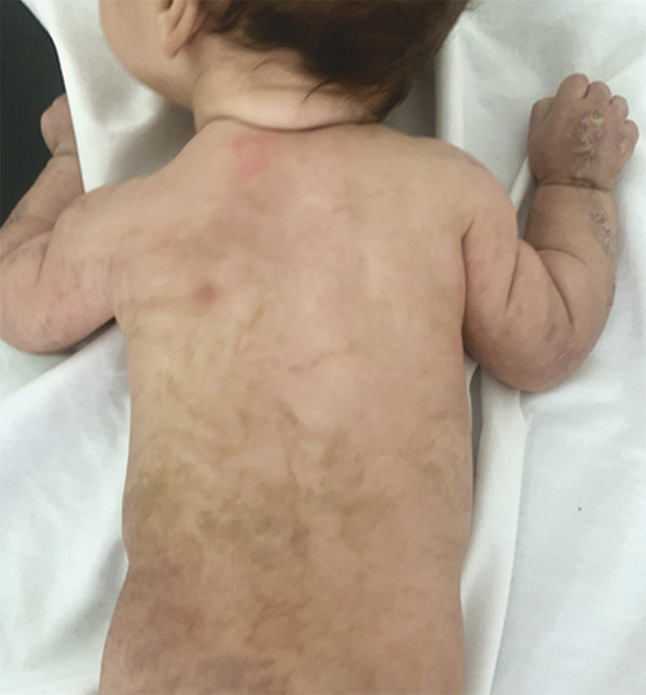
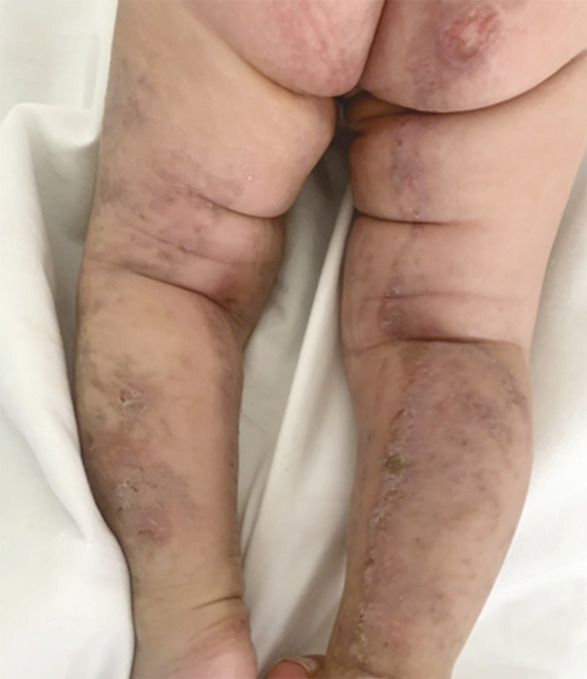
Ce nourrisson de 3 mois, de sexe féminin, issu d’un mariage consanguin et dont la mère rapportait des antécédents d’avortements spontanés, a été adressé pour des lésions vésiculo-croûteuses des membres évoluant depuis sa naissance (fig. 1 ). Cliniquement, il existait une pigmentation brune linéaire du tronc (fig. 2 ) ainsi que des papules verruqueuses des bras et des jambes (fig. 3 ), suivant les lignes de Blaschko*.
La biopsie cutanée était en faveur d’une incontinentia pigmenti au stade papulo-kératosique. Les examens ophtalmologique et neurologique étaient normaux et il existait une hyperéosinophilie. Le diagnostic d’incontinentia pigmenti a été retenu sur l’association de deux critères majeurs, selon les critères actualisés de Landy et Donnai – éruption néonatale et signes cutanés typiques –, à deux critères mineurs : hyperéosinophilie et histologie cutanée caractéristique. La prise en charge a été fondée sur des soins locaux (antiseptiques, émollients) et la photoprotection avec un suivi dermatologique, neurologique, ophtalmologique et dentaire codifié.
La biopsie cutanée était en faveur d’une incontinentia pigmenti au stade papulo-kératosique. Les examens ophtalmologique et neurologique étaient normaux et il existait une hyperéosinophilie. Le diagnostic d’incontinentia pigmenti a été retenu sur l’association de deux critères majeurs, selon les critères actualisés de Landy et Donnai – éruption néonatale et signes cutanés typiques –, à deux critères mineurs : hyperéosinophilie et histologie cutanée caractéristique. La prise en charge a été fondée sur des soins locaux (antiseptiques, émollients) et la photoprotection avec un suivi dermatologique, neurologique, ophtalmologique et dentaire codifié.
L’incontinentia pigmenti est une maladie rare dont l’incidence est estimée à 0,7 cas pour 100 000 naissances.1 Il s’agit d’une dysplasie ectodermique multisystémique liée au chromosome X, due à des mutations héréditaires ou sporadiques de novo (plus de 75 %) du gène de l’inhibiteur de la sous-unité gamma du facteur nucléaire kappa B kinase (IKBKG/NEMO).2 L’atteinte cutanée typique permet de reconnaître cette affection. Quatre stades sont bien caractérisés : vésiculo-bulleux, verruqueux, hyperpigmenté et atrophique/hypopigmenté. Des anomalies neurologiques, oculaires et dentaires peuvent s’y associer, qui imposent un dépistage et une prise en charge adaptée.
* NDLR : lignes épidermiques décrites à partir de dermatoses linéaires congénitales, correspondant peut-être au trajet de migration de cellules épidermiques embryonnaires en direction antérolatérale à partir de la crête neurale. Source : dictionnaire de l’Académie de médecine 2022.
Références
1. Bodemer C, Diociaiuti A, Hadj-Rabia S, Robert MP, Desguerre I, Manière MC, et al. Multidisciplinary consensus recommendations from a European network for the diagnosis and practical management of patients with incontinentiapigmenti. J Eur Acad Dermatol Venereol 2020;34:1415-24.
2. Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic rearrangement in NEMO impairs NF-kappa B activation and is a cause of incontinentia pigmenti. The International incontinentia pigmenti (IP) consortium. Nature 2000; 405:466-72.
2. Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic rearrangement in NEMO impairs NF-kappa B activation and is a cause of incontinentia pigmenti. The International incontinentia pigmenti (IP) consortium. Nature 2000; 405:466-72.
Une question, un commentaire ?
Sur le même thème
Article Web
Exercice
Article Web