Vous recevez dans la soirée aux urgences un homme de 45 ans pour des anomalies de sa prise de sang. C’est un homme sans antécédent notable.
Depuis trois jours, il présente des gingivorragies et des épistaxis récidivantes bilatérales. Votre examen clinique retrouve un purpura des membres inférieurs.
Il arrive avec un hémogramme fait la veille par son médecin traitant, qui retrouve :
Question 1 : Quel(s) est(sont) les examens à demander aux urgences dans un premier temps devant cette bicytopénie ?
Toujours demander un frottis sanguin devant une bicytopénie : recherche des schizocytes, des cellules anormales (blastes…)
Devant une thrombopénie : éliminer une CIVD
Examen à réaliser en cas d’anémie microcytaire, ici ce n’est pas le cas
L’anémie par carence en B9/B12 est non régénérative et macrocytaire
En première intention, pour savoir si la bicytopénie est centrale ou périphérique
Devant une bicytopénie : distinguer l’origine centrale de l’origine périphérique par le dosage des réticulocytes et l’examen du frottis sanguin.
Causes périphériques de bicytopénie :
- syndrome de microangiopathie thrombotique (MAT) = hémolyse mécanique ;
- syndrome d’Evans (thrombopénie et anémie hémolytique auto-immune).
Causes centrales : nécessite alors une exploration médullaire
°Moelle riche :
- leucémie aigüe, SMD, SMP ;
- carence vitaminique (B9, B12) : peu probable ici car l’anémie est normocytaire.
°Moelle pauvre :
- aplasie médullaire ;
- myélofibrose
Le bilan aux urgences est le suivant :
GB=3 G/L, dont PNN=300/mm3, le reste de la formule est en attente, Hb = 7 g/dL, VGM = 86 fL, plaquettes = 10 G/L, réticulocytes = 20 G/L.
Absence de schizocytes.
Question 2 : Quelle(s) est(sont) alors vos hypothèses possibles à ce stade ?
Classiquement, il s’agit d’une bicytopénie périphérique, donc régénérative
Se révèle par une mono-/bi-/pancytopénie d’origine centrale. Le taux de GB peut être normal (absence de lymphopénie, ici nous n’avions pas le reste de la formule)
Peu probable ici car l’anémie est normocytaire
Pancytopénie d’origine centrale
La NFS retrouve ici une pancytopénie (car PNN < 1 000/mm3, anémie et thrombopénie) d’origine centrale, car réticulocytes < 120 G/L.
Évoquer les causes de pancytopénies à « moelle riche » :
- hémopathies surtout myéloïdes (LA, syndrome myélodysplasique), et parfois lymphoïdes (LLC avec envahissement médullaire par ex) ;
- carences vitaminiques avancées (B9, B12) peuvent entraîner une pancytopénie. La carence en B12 donne une anémie macrocytaire ;
- envahissement médullaire par une métastase d’un cancer solide.
Pancytopénie à « moelle pauvre » au myélogramme : imposant de faire une BOM :
- myélofibrose primitive ;
- aplasie médullaire ;
- syndrome d’activation macrophagique.
Le complément du bilan retrouve le bilan d’hémostase suivant : TP = 28 %, TCA = 60 secondes pour un témoin à 23 sec, fibrinogène = 0,8 g/L.
Vous suspectez donc une coagulopathie intravasculaire disséminée (CIVD).
Question 3 : Quel(s) examen(s) demandez-vous pour aller dans ce sens ?
CIVD = consommation de tous les facteurs de la coagulation. Le dosage du facteur V permet d’éliminer une carence en vitamine K
Facteurs de la voie endogène. N’entraîne qu’un allongement du TCA, sans modification du TP en cas de déficit
Le tableau n’est pas évocateur d’une maladie de Willebrand. Examen sans intérêt pour le diagnostic de CIVD
Les D-dimères font partie des produits de dégradation de la fibrine et sont élevés en cas de CIVD
CIVD = coagulopathie intravasculaire disséminée :
- consommation des facteurs de la coagulation de la voie exogène et endogène : Baisse du TP et allongement du TCK ;
- consommation du fibrinogène ;
- thrombopénie de consommation (ici la thrombopénie est probablement aussi liée à l’hémopathie) ;
- augmentation des produits de dégradation de la fibrine (PDF : D-dimères+++, complexes solubles, monomère de fibrine : non validé).
Le complément de l’hémogramme est le suivant : GB = 3 G/L, dont PNN = 300/mm3, lymphocytes = 1 700/mm3, blastes = 1 000/mm3, PNéo = 0/mm3, PNBaso = 0/mm3, Hb = 7 g/dL, VGM = 86 fL, plaquettes = 10 G/L, réticulocytes = 20 G/L.
Question 4 : Quelle(s) est(sont) alors vos hypothèses possibles avec ces nouvelles données ?
Pancytopénie d’origine centrale sans excès de blastes ou cellules anormales
Cytopénies d’origine centrale sans excès de blastes ou cellules anormales
Il est possible d’avoir une circulation de blastes dans la myélofibrose primitive mais avec une myélémie
Question 5 : Quel(s) examen(s) demandez-vous à ce stade ?
Dans un premier temps : toujours faire un myélogramme, la BOM sera faite en cas de myélogramme non contributif
Un myélogramme par voie sternal est réalisable quel que soit le bilan d’hémostase et le chiffre de plaquettes
Dans les hypothèses diagnostiques, examen pas nécessaire
L’exploration médullaire est indispensable au diagnostic de leucémie aiguë et permet d’affiner le diagnostic par les examens cytogénétique et moléculaire
Myélogramme : analyse cytologique des cellules de la moelle osseuse.
Se fait sous anesthésie locale chez l’adulte, après asepsie.
Deux voies possibles : voie sternale, quel que soit le chiffre de plaquettes. La voie iliaque est plus à risque d’hématome et nécessite donc un contrôle de l’hémostase avec si besoin transfusion de plaquettes et de PFC dans ce cas-là.
Un myélogramme est finalement réalisé le lendemain matin.
La conclusion est la suivante : moelle riche, envahie par 80 % de blastes granuleux de taille moyenne à grande, marquant la MPO. Certains possèdent un corps d’Auer dans leur cytoplasme.
Absence de mégacaryocyte vu sur le frottis. Le reste des cellules ne présentent pas d’anomalie morphologique.
Question 6 : Quel(s) est(sont) alors les diagnostics possibles ?
Ici il s’agit d’une leucémie aiguë myéloïde, donc seule proposition vraisemblable
Le myélogramme retrouve classiquement une moelle pauvre
Blastes ici granuleux et marquant la MPO donc blastes myéloïdes
Absence de signes de dysplasies sur les cellules restantes, antécédents de NFS normale
Absence d’argument pour un SMP sous-jacent : NFS antérieure normale
- il s’agit ici d’un diagnostic de leucémie aiguë car blastes médullaires > 20 % ;
- blastes myéloïdes car : granuleux, marquant la myélopéroxydase (examen d’immunochimie sur la moelle – pas fait systématiquement si le caractère granuleux est cytologiquement sûr). La présence d’un corps d’Auer se voit dans les blastes myéloïdes (les corps d’Auer correspondent à des débris nucléaires) ;
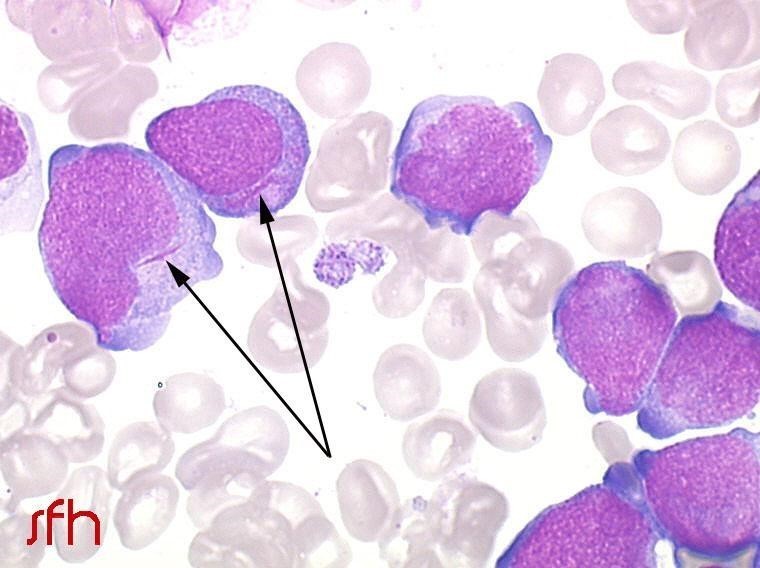
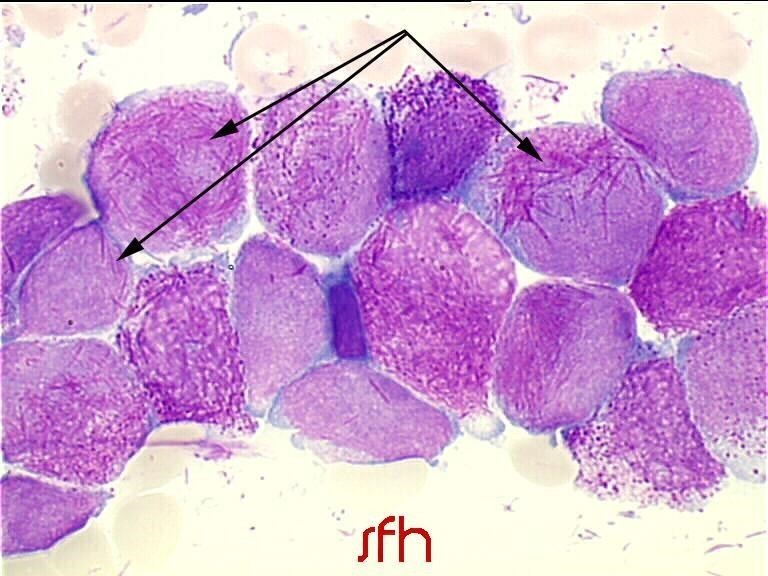
- dans la LAM 3 : on observe classiquement des corps d’Auer nombreux et disposés en fagot. Image 1 - LAM 2 : blastes granuleux avec présence d’un corps d’Auer unique dans le cytoplasme. Copyright : SFH Image 2 - LAM 3 : présence dans le cytoplasme de nombreux corps d’Auer, en fagot. Copyright : SFH
CLASSIFICATION OMS 2016 DES LAM (pour information)
1) LAM avec anomalies cytogénétiques récurrentes :
LAM t(8;21)
LAM avec inv(16), t(16;16)
LAM avec t(15;17) = LAM3
LAM avec t(9;11)
LAM avec t(6;9)
LAM avec inv(3)
LAM avec mutation NPM1, mutation double mutant CEBPA
2) LAM avec dysplasies sous-jacentes
Au moins 50 % de cellules dysplasiques
Antécédent de SMD connu
Anomalie cytogénétique de SMD
3) LAM secondaires à un traitement
4) LAM sans autres spécificités
LAM classification FAB non classables dans les autres items
5) Sarcome myéloïde
6) Prolifération myéloïdes associées au syndrome de Down
Il est maintenant hospitalisé dans votre service d’hématologie pour la prise en charge de sa LAM4. Les constantes sont les suivantes : T° 37,2, FC = 74 batt/min, PA = 120/78. Votre examen clinique retrouve uniquement ce purpura des deux jambes. Il persiste depuis la veille une épistaxis peu abondante ; malgré un méchage antérieur. Le reste de l’examen physique est strictement normal. Il n’y a pas de syndrome tumoral périphérique.
Question 7 : Quelles sont vos mesures de prise en charge immédiate en attendant le début de la chimiothérapie ?
Pas d’antibiothérapie ici car le patient est apyrétique, pas de foyer infectieux à l’examen physique
À proposer systématiquement chez l’homme jeune. Le diagnostic de LAM n’est pas une contre-indication
Ne peut être fait avant le contrôle des troubles de l’hémostase
Seuil lors d’une CIVD plaquettes > 50 G/L
Pour maintenir le fibrinogène > 1 g/L
Prise en charge des complications d’une leucémie aiguë au diagnostic :
- syndrome hémorragique :
CIVD : transfusion de PFC (plutôt que de fibrinogène) : objectif TP > 50 %, fibrinogène > 1 g/L, traitement étiologique ++ ;
transfusion plaquettes si < 20 G/L ou si syndrome hémorragique et/ou CIVD : < 50 G/L ;
- syndrome anémique :
transfusion de CGR selon la tolérance de l’anémie, seuil < 8 g/dL (10 g/dL chez le patient coronarien), prudence si LA hyperleucocytaire ;
- problème infectieux :
ATB large spectre IV couvrant Pseudomonas aeruginosa si fièvre ;
- leucostase : dans les LAM hyperleucocytaires (> 50 G/L de GB) : atteinte pulmonaire surtout et cérébrale. Indication à une cytoréduction en urgence et indication des CTC ;
- syndrome de lyse tumoral : LA hyperleucocytaire ++.
Mise en condition du malade :
- hospitalisation dans un secteur stérile/chambre à flux/isolement protecteur pour prévention du risque infectieux (aspergillaire) ;
- pose d’une voie veineuse centrale (plutôt KT central dans les LA que pose de PAC : facilité à retirer en cas de complications infectieuses) après transfusion si besoin et une fois une éventuelle CIVD contrôlée ;
- bilan préthérapeutique :
- sérologies ;
- bilan hépatique/rénal ;
- ETT pour évaluation de la FEVG (avant anthracyclines surtout) ;
- CECOS si possible chez l’homme jeune.
Question 8 : Quels éléments sont nécessaires pour discuter de son traitement en réunion de concertation pluridisciplinaire ?
Pas nécessaire dans les leucémies aiguës
Uniquement en cas de point d’appel clinique d’une atteinte neuroméningée
Le score OMS est toujours un facteur pronostique dans les hémopathies
Éléments pronostiques dans la prise en charge des LAM :
pour la prise en charge ultérieure : obtention de la rémission complète après induction. Mesure de la maladie résiduelle (MRD) en biologie moléculaire.
l’âge (> 65 ans) et l’état général du malade (score OMS) ;
biologie moléculaire (= analyse des gènes) : technique plus fine pour rechercher d’autres marqueurs pronostiques et permettent le suivi ;
analyse du caryotype (= analyse des chromosomes) : recherche d’anomalies cytogénétiques acquises (c’est-à-dire qu’elles ne sont pas constitutionnelles, mais ne sont présentes que dans les cellules hématopoïétiques : on dit qu’elles sont clonales), si besoin compléter par de la FISH ;
Atteinte neuroméningée :
- moins de 5 % des LAM (plus fréquent dans les LAL : dépistage et prophylaxie systématique dans ce cas) ;
- se recherche uniquement si signes cliniques en faveur d’une atteinte neuroméningée : hypoesthésie de la houppe du menton (toujours à rechercher lors de l’examen clinique), ou tout autre signe de localisation ;
- situations particulières où la PL se discute : les LAM hyperleucocytaires (mais non consensuelles).
La RCP propose une chimiothérapie d’induction par anthracycline et cytarabine. Le reste de la prescription est la suivante : sérum physiologique : 4 000 mL/j, prophylaxies infectieuses : amoxicilline, posaconazole, valaciclovir. Traitements antiémétiques.
Après trois jours de chimiothérapie, l’infirmière vous appelle car il se sent essoufflé et présente une orthopnée depuis le matin.
Les constantes sont les suivantes : T° = 37,9, FC=75 batt/min, PA = 150/85, FR = 25/min, SatO2 = 89 % en air ambiant.
Le bilan du jour retrouve une persistance de la CIVD avec une thrombopénie à 45 G/L, mais le syndrome hémorragique s’est amendé.
Question 9 : Quelle(s) est(sont) la/les hypothèse(s) qui vous semble(nt) probable pour expliquer le tableau respiratoire ?
Toujours penser à la cause infectieuse chez le patient neutropénique
La leucostase se voit chez les patients hyperleucocytaires
Ce sont les anthracyclines qui donnent des toxicités cardiaques
Du fait du contexte
Complications possibles compte tenu de la CIVD et de la thrombopénie
Finalement, l’évolution est favorable après déplétion hydrosodée. La radiographie de thorax faite au lit ne retrouvait pas d’argument pour un foyer infectieux.
Trois jours plus tard, lors de votre visite, les constantes sont les suivantes : T° = 39,4 °C, PA = 114/74, FC = 110 batt/min, SatO2 = 100 %, FR = 15/min.
Vous ne trouvez pas de point d’appel infectieux clinique lors de votre examen. Il ne présente pas de marbrures ou de cyanose et la diurèse est conservée. Son cathéter posé après correction de la CIVD n’est pas inflammatoire.
Sa NFS du jour retrouve : GB = 0,2 G/L, Hb = 10 g/dL, plaquettes = 20 G/L.
Question 10 : Quelle(s) est(sont) la(les) proposition(s) exacte(s) concernant les examens à demander à l’infirmière ?
La documentation microbiologique est importante
Les prélèvements ne doivent pas retarder le début des antibiotiques, une seule paire d’hémocultures, puis les autres seront faites après
Toujours prélever les corps étrangers, avec recherche d’un différentiel de pousse
Marqueur indirect d’infection fongique invasive
Question 11 : Quelle est votre prise en charge de cette complication ?
Indication des glycopeptides : choc septique, portage de SARM connu, point d’appel cutané (KT purulent ou inflammatoire, lésions cutanées comme un furoncle, mucite érosive)
En cas de fièvre persistante au-delà de 5 jours
Ne s’utilise pas dans le traitement de l’aplasie fébrile
Seulement si argument pour infection de KT ou choc septique
L’infirmière vous appelle car votre malade se met à frissonner brutalement au moment de la réalisation de l’hémoculture. Les constantes sont alors les suivantes : PA = 75/40, FC = 140 batt/min, T = 39, SatO2 = 99 %. Après remplissage par 500 mL de sérum salé isotonique, la tension reste à 80/50.
À l’examen, votre patient est marbré.
Question 12 : Quelle est alors votre prise en charge ?
Tableau de choc septique, donc indication d’élargir d’emblée le spectre
Les infections fongiques ne sont pas responsables de choc septique
Ne s’utilise pas dans le traitement de l’aplasie fébrile
Car tableau de choc septique
Résumé de la prise en charge d’une aplasie fébrile (durée > 7 jours) :
- germes responsables : BGN : 1/3 (E. coli ++, Pseudomonas), CGP : 2/3 (staphylocoques, streptocoques).
1) Évaluer la gravité : Recherche de signes de choc/sepsis sévère.
2) Recherche d’une porte d’entrée : examen clinique approfondi +++ (KT, périnée +++) + Atcd patients (Porteur BLSE/SARM +++).
3) Prélèvements infectieux : hémocultures KT + périph en même temps, ECBU (pas de BU), radio de thorax, agnémie galactomannane.
4) Traitement sans attendre les résultats :
-Bêtalactamine large spectre (couverture BGN dont le Pyo et CGP) :
- pipéracilline + tazobactam // C3G large spectre en monothérapie en première intention ;
- signes de sepsis sévère : ajout aminosides ;
- signes de porte d’entrée cutanée franche, ou patient colonisé à SARM : ajout d’un glycopeptide ;
- si patient BLSE connu : ATB orientée.
5) Réévaluation ++++ clinique et biologique (documentation ou non)
Persistance de la fièvre à 5 jours (et neutropénie > 7 jours) : ajout antifongique.
Arrêt des ATB : après la sortie d’aplasie et pas avant.
La fièvre persiste malgré les antibiotiques. Cela fait maintenant 5 jours, et vous décidez donc de réaliser un scanner thoracique dont les images sont les suivantes.
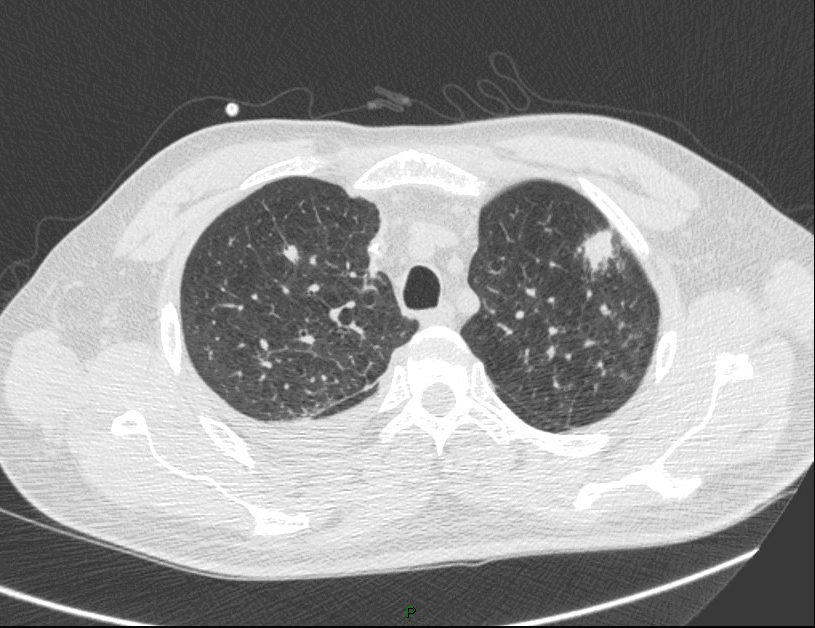
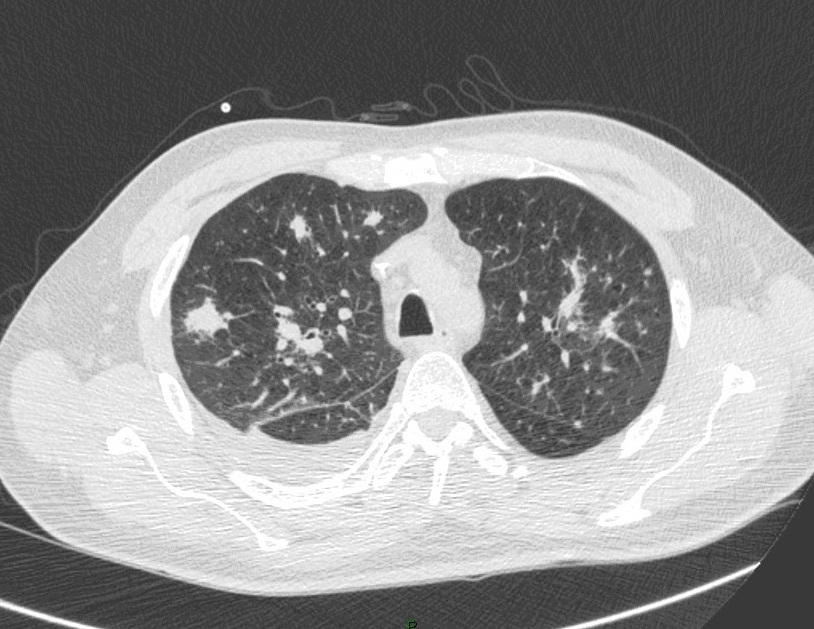
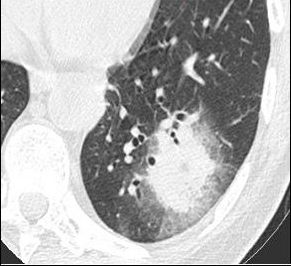
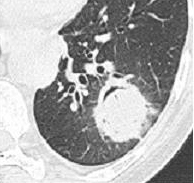
Question 13 : Comment décrivez-vous ces images ?
C’est l’image qui est retrouvée dans l’aspergillome : au sein d’une caverne (souvent séquellaire de tuberculose) présence d’un nodule aspergillaire
Nodule pulmonaire entouré de verre dépoli
Présence d’un épanchement pleural gauche
Quesyion 14 : Quelle(s) est(sont) alors les hypothèses diagnostiques devant ces images ?
Hypothèse la plus probable devant le terrain de neutropénie prolongée et les images en faveur
On verrait plutôt des images avec le signe du grelot
Absence de condensations alvéolaire ou de bronchogramme aérien
Les images sont peu en faveur : l’image typique étant la caverne de l’apex pulmonaire. La tuberculose pulmonaire donne rarement des images nodulaires
Impossible de voir une embolie pulmonaire sur des coupes parenchymateuses
Aspergillose pulmonaire invasive = infection fongique invasive la plus fréquente chez le patient avec neutropénie prolongée (> 7 jours de neutropénie).
Tableau clinique classique :
- fièvre persistante sous antibiotiques, souvent bien tolérée, sans signe de sepsis ;
- symptômes respiratoires selon l’atteinte (toux, dyspnée, hémoptysie) ;
- imagerie :
°précoce : signe du halo : nodule unique ou multiple, avec verre dépoli autour (signe du grelot se voit dans l’aspergillome compliquant une caverne tuberculeuse par exemple) ;
°plus tardif : condensation non spécifique, puis apparition d’un croissant gazeux traduisant la nécrose du parenchyme.
- éléments diagnostiques :
°test biologique : antigénémie galactomannane, bêta-D-glucane ;
°prélèvement respiratoire : analyse mycologique d’un ECBC (souvent peu rentable), fibroscopie bronchique avec LBA.
- Traitement :
°antifongiques systémiques : azolés ++ (voriconazole), amphotéricine B liposomale ;
°dans tous les cas, quel que soit le résultat du LBA : neutropénie fébrile persistante au-delà de 5 jours : traitement antifongique empirique jusqu’à la sortie d’aplasie.
Il est maintenant en rémission complète, et son infection fongique invasive est traitée. Du fait de marqueurs génétiques de mauvais pronostic, il a été décidé en RCP la réalisation d’une allogreffe en consolidation.
Sa femme vous demande de réaliser un typage chez elle car elle souhaite lui donner sa moelle osseuse et pense être compatible car ils ont le même groupe sanguin.
Question 14 : Que lui répondez-vous ?
Si elle souhaite donner sa moelle, elle doit s’inscrire sur le fichier international, et son typage sera fait lors de l’inscription (auprès de l’EFS)
Il s’agit uniquement des frères et sœurs
C’est la compatibilité HLA qui est nécessaire
Principe de l’allogreffe :
- donneur = celui qui donne sa moelle osseuse, receveur = le patient ;
- but : traitement de consolidation des leucémies aiguës surtout ;
- deux effets : GVL (Graft versus leukemia : effet du greffon contre les cellules résiduelles de la leucémie : effet recherché), GVH (graft versus host : lymphocytes T du donneur reconnaissent les cellules du receveur comme étrangère et l’attaquent) ;
- après un conditionnement (chimiothérapie pour mettre en aplasie et « détruire » le système immunitaire du receveur) ;
- recueil des cellules souches hématopoïétiques du donneur soit par prélèvement de moelle osseuse au bloc sous AG, soit après mobilisation par G-CSF et cytaphérèse ;
- réinjection des cellules du donneur (se passe comme une transfusion) ;
- traitement immunosuppresseur pour éviter la GVH (durée variable).
Les frères et sœurs du patient ont une chance sur 4 d’être compatible : transmission du système HLA de façon codominante : on hérite un haplotype paternel et un haplotype maternel : donc probabilité d’être identique de 1 sur 4.
En dehors des frères et sœurs : la probabilité de sa femme d’être compatible avec le patient est la même que la population générale.
En absence de donneur dans la famille : on interroge le fichier international (et non national) de don de moelle : fichier anonyme.
On ne respecte pas toujours la compatibilité ABO lors du don de moelle, c’est la compatibilité HLA qui est nécessaire.
À noter qu’il se développe des greffes alternatives avec des donneurs moins compatibles : donneur fichier de compatibilité moindre, ou des greffes dites « haplo-identiques » : avec un seul haplotype en commun entre le donneur et le receveur (de ce fait, les parents peuvent donner) : mais il s’agit encore de phase expérimentale, donc pas recommandé.


Causes périphériques de bicytopénie :
- syndrome de microangiopathie thrombotique (MAT) = hémolyse mécanique ;
- syndrome d’Evans (thrombopénie et anémie hémolytique auto-immune).
Causes centrales : nécessite alors une exploration médullaire
°Moelle riche :
- leucémie aigüe, SMD, SMP ;
- carence vitaminique (B9, B12) : peu probable ici car l’anémie est normocytaire.
°Moelle pauvre :
- aplasie médullaire ;
- myélofibrose